Microbime:微生物组学领域的标准制定
简介

标题:Developing standards for the microbiome field
微生物组学领域的标准制定
杂志:《Microbiome》
影响因子:11.607
发表时间:2020年6月26日
解读:章小鱼
编辑:很跩的土豆
热心肠日报
https://www.mr-gut.cn/papers/read/1044216095
为微生物组分析定制的标准DNA样本和评估框架
创作:unannotated 审核:刘永鑫-中科院-宏基因组
由于缺乏公认的参考菌群DNA,微生物组科学家难以评估不同实验方法和分析流程所带来的偏差;
本研究开发了两种参考菌群DNA,包含20种常见的肠道微生物菌株,涉及两种不同比例的菌株组合;
对参考菌群DNA进行全基因组和16S测序并分析,发现不同工具可带来差异,指标包括敏感性、假阳性丰度、多样性和相似性;
作为WHO的国际微生物组研究标准候选,本文开发的参考菌群DNA将在多个实验室进行评估,以促进微生物领域研究的标准制定。
主编推荐语:本研究中,作者开发了NIBSC参考菌群DNA Gut-Mix-RR和Gut-HiLo-RR,以及用于评估生物信息学工具和流程偏差的四项措施框架。使用这些参考菌群和报告系统,作者通过分析由Gut-Mix-RR和Gut-HiLo-RR生成的宏基因组测序和16S rRNA基因测序数据,对各种生物信息学工具进行了独立评估,并证明大多数生物信息学工具极大地夸大了微生物组健康的关键指标,例如多样性估计。在所有测试的工具中,都存在偏差,最终数据集中的敏感性和假阳性的相对丰度之间存在明显的权衡。最终展望未来,作者建议领域内使用高度复杂的特定于本领域的菌群,以确保流程基准测试适用于特定目的。
正文
特色:(1)为微生物组学分析制备了参考菌群DNA;(2)建立了评估体系。
导读:微生物组学已经成立了十多年,但尚无公认的统一分析标准。微生物组学分析方法的标准化对于整个微生物组研究有重要意义。在这篇文章中,作者阐述了由国家生物标准和控制研究所(National Institute for Biological Standards and Control,NIBSC)基于二代测序研发的微生物组学分析的参考菌群DNA:Gut-Mix-RR和Gut-HiLo-RR,以及评估生物信息学工具和流程偏差的四个参数,它们将成为WHO国际参考菌群DNA,并有望成为全球工作标准。
背景:二代测序技术(next-generation sequencing, NGS)的进步促进了微生物组学的快速发展。随着测序成本降低,基于测序的研究方法可用于更多的科研领域,如NGS技术已经应用于人群微生物组学研究。然而不同方法之间的差异导致微生物组学研究的最佳方法尚不确定,这其中包括样品存储和收集的差异、不同DNA提取过程的差异、不同NGS平台的差异、使用16S扩增子方法可以扩增不同16S rRNA基因区域以及实验室偏好和生物信息学流程差异,因此开发出有效的参考试剂()对于微生物组学研究的标准化是非常有必要的。鉴于微生物组学研究方法的多步骤性和复杂性,可能需要一系列的参考试剂和有效的报告体系来标化各步骤产生的差异并评估捕获已知和未知微生物丰度的准确性。
结果:
1. 开发参考试剂和报告系统
有效标准化微生物组学研究方法至少需要三种参考试剂,分别为控制文库制备、测序和生物信息学流程中偏差的DNA试剂,控制DNA提取中偏差的全细胞试剂,控制抑制剂或储存条件偏差的加有基质的全细胞试剂。在这项研究中,作者开发了DNA参考试剂用于下游分析的标准化,他们创建了两个DNA模拟菌群(即DNA参考试剂),分别为Gut-Mix-RR和Gut-HiLo-RR,由20种常见肠道微生物均匀交错组成(表1),包括5个门、13个科、16个属和19个种的菌株,以允许测试不同分类标准下检测流程的性能。参考试剂的一个关键成分是报告系统(可重复报告检测结果),研究者设置了内部报告系统来评估下游微生物组学分析。
在开发报告系统时,研究者考虑了能够捕获分析流程中常见偏倚并反映微生物组研究报告结果的参数,为了保证报告系统适用于大多数研究,他们最终选用了敏感性、假阳性相对丰度(false positive relative abundance,FPRA)、多样性、相似度,分别来衡量流程检测样品中已知物种的能力、流程如何引入假阳性物种、流程中总物种数和物种组成。
表1 NIBSC Gut-Mix-RR和Gut-HiLo-RR的菌株和特征

Gut-Mix-RR(%)和Gut-HiLo-RR(%),基于基因组拷贝的相对数量;GC-content(%),基于原始物种描述中可用或不可用(*)的基因序列;Accession numbers,GeneBank Accession numbers或RefSeq accession numbers;NC_008530,加式乳杆菌(Lactobacillus gasseri)ATCC 33323的RefSeq accession。该研究中所有序列来自于NCBI Bioproject ID PRJNA622674;16S拷贝数和序列数,基于IMG/M序列分析。
2. 使用NOBSC RR评估宏基因组测序的生物分类学
很少有研究单独比较分析宏基因组测序数据的生物信息学工具。因此为了验证NIBSC-Gut-Mix-RR、NIBSC-Gut-HiLo-RR和报告系统的适用性,他们研究了生物信息学工具之间的结果变异性。在5个重复样品上用参考试剂进行Shallow shotgun测序,并使用五种常见的生物信息学工具(MetaPhAn2、Kraken、Bracken、Kaiju和Centrifuge)进行分析。结果表明在种水平,两种参考试剂的生物信息学工具所有报告值都存在显著差异(图1)。灵敏度和FPRA之间存在显著平衡,敏感性最低的工具Kaiju具有最高的FPRA,而FPRA最低的工具 MetaPhlan2具有较低的敏感性。对于Gut-Mix-RR,在五种生物信息学工具中,敏感性从73%到100%,其中Kaiju是唯一可检测试剂中所有菌种的生物信息学工具,这主要是由于青枯病菌(Blautia wexlerae)和古鲁球菌(Ruminococcus gauverauii)的检测问题;Kraken,Bracken,Centrifuge在属水平归类正确,而在种水平就不准确了;MetaPhlAn2的问题是无法检测到某个菌属中的任何菌种。MetaPhlAn2是唯一FPRA为0%的生物信息学工具,而有多种工具却无法给多种低丰度菌种归类。例如,当只有两种拟杆菌种出现时,Kraken和相关的Bracken都检测到13种不同的拟杆菌种;而Centrifuge则将大量埃希氏菌属分到志贺氏菌属,Kaiju则检测到许多与参考试剂无关的低丰度菌种。各个工具的敏感性和FPRA的差异导致菌种多样性和相似性的差异,其中多样性可相差9倍。MetaPhlAn2具有最佳的多样性预测能力,而Kaiju和MetaPhlAn2与Gut-Mix-RR的实际成分相似度最高。
接下来,作者使用Gut-HiLo试剂评估了相同生物信息学工具的性能,该试剂挑战了检测低丰度菌株的能力(图1)。广义上讲,每种工具的性能都与Gut-Mix相似,其中MetaPhlAn2是唯一具有0%FPRA的工具,而Kaiju是唯一可以检测试剂中所有菌种的工具。对于Centrifuge和MetaPhlAn2的敏感性降低,Kaiju和MetaPhlAn2仍具有最高相似度。

图1 使用NIBSC Gut-Mix-RR和Gut-HiLo-RR在种水平和属水平上比较不同生物信息学工具的性能。A 和已知成分的试剂相比,Gut-Mix-RR利用5种不同的宏基因组分类工具计算出的每个菌种的相对丰度 B 和已知成分的试剂相比,Gut-HiLo-RR利用5种不同的宏基因组分类工具计算出的每个菌种的相对丰度 C 和已知成分的试剂相比,Gut-Mix-RR利用5种不同的宏基因组分类分析工具以及16S rRNA分类工具计算出的每个菌种的相对丰度 D和已知成分的试剂相比,Gut-HiLo-RR利用5种不同的宏基因组分类工具以及16S rRNA分类工具计算出的每个菌种的相对丰度 E使用Gut-Mix-RR评估用于计算菌种流程性能的报告值 F使用Gut-HiLo-RR评估的用于计算菌种通道性能的报告值 G使用Gut-Mix-RR评估用于计算属的通道性能的报告值 H使用Gut-HiLo-RR评估用于计算菌属通道性能的报告值 Mp MetaPhlAn2,Kj Kaiju,Kr Kraken,Br Bracken,Cn Centrifuge,Sens敏感性,FPRA假阳性相对丰度,Div多样性,Sim相似性
为了研究生物信息学工具之间的差异是否可能导致不同的微生物群落分析结果,他们利用NIBSC试剂对五个市售的合成菌群进行重复分析(图2)。可视化结果显示菌群分组受参考试剂影响更大,而非生物信息学工具(图2)。群落组成方差的分析结果表明,参考试剂产生了数据中最大的变异,而生物信息学工具则仅解释了约5.29%的变化。这表明,尽管信息学工具的选择会显著影响菌种组成,但似乎并未改变微生物群落之间的潜在关系。随后他们使用4种报告系统综合评估流程性能并发现生物信息学工具主要影响alpha多样性。这表明不同报告系统可以准确评估微生物组研究结果的不同方面,同时说明了参考试剂要用多种报告系统的重要性。
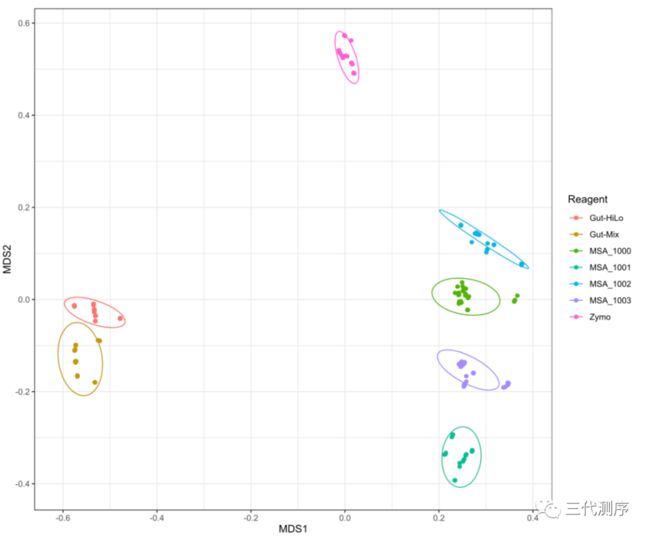
图2 通过各种方法在测序和分类分析后可视化不同模拟菌落之间的关系。5种参考试剂经过shotgun测序和5种生物信息学工具MetaPhlAn2,Kaiju,Kraken,Bracken和Centrifuge进行分类分析后,构建的Bray-Curtis异质性矩阵的nMDS图。Gut-HiLo = NIBSC Gut-HiLo-RR,Gut-Mix = NIBSC Gut-Mix-RR,MSA_1000 = ATCC MSA-1000,MSA_1001 = ATCC MSA-1001,MSA_1002 = ATCC MSA-1002,MSA_1003 = ATCC MSA-1003,Zymo = ZymoBIOMICS微生物群落标准
3. 观察菌种组成对通道性能的影响
目前尚无公认的参考试剂,也没有文献明确指出什么才是合适的微生物参考试剂以及不同试剂成分如何影响基准研究或不同微生物组成如何影响不同通道的准确性。为了了解这些因素如何影响微生物组领域的标准化,他们比较了5种市售参考试剂有效基准化生物信息学工具的能力。使用这些商业模拟菌落的序列,他们计算了5种生物信息学工具的敏感性、FPRA和相似性,然后与两种NIBSC参考试剂的测量值进行比较。在所有工具中,敏感性和FPRA会受参考试剂的影响,这说明了在改变目标样品的微生物组成时生物分类工具性能受到影响(图3)。生物分类工具的敏感性也因试剂的菌种数量和菌种组成而有明显的变化趋势(图3)。在所有工具中,较高菌种丰度和波动菌种丰度的试剂有较低的敏感性,而均匀菌群丰度的试剂有较高的敏感性(图3)。但是即使考虑了菌种组成和数量,与具有相似菌株数量和组成的其他试剂相比,4种生物信息学工具对NIBSC试剂均具有较低的敏感性。这表明菌种组成也会影响测序和生物信息学工具的性能,这可能是由于GC含量偏差或它们是否存在于公共数据库而导致。参考试剂在不同通道中的相似性也有很大差异。然而变异通常是针对特定通道的,没有清晰的模式出现。总的来说不同的参考试剂在不同的通道上会给出不同的结果。因此有效的生物信息学工具的基准测试需要能够模拟目标样品可能物种组成的特定试剂。
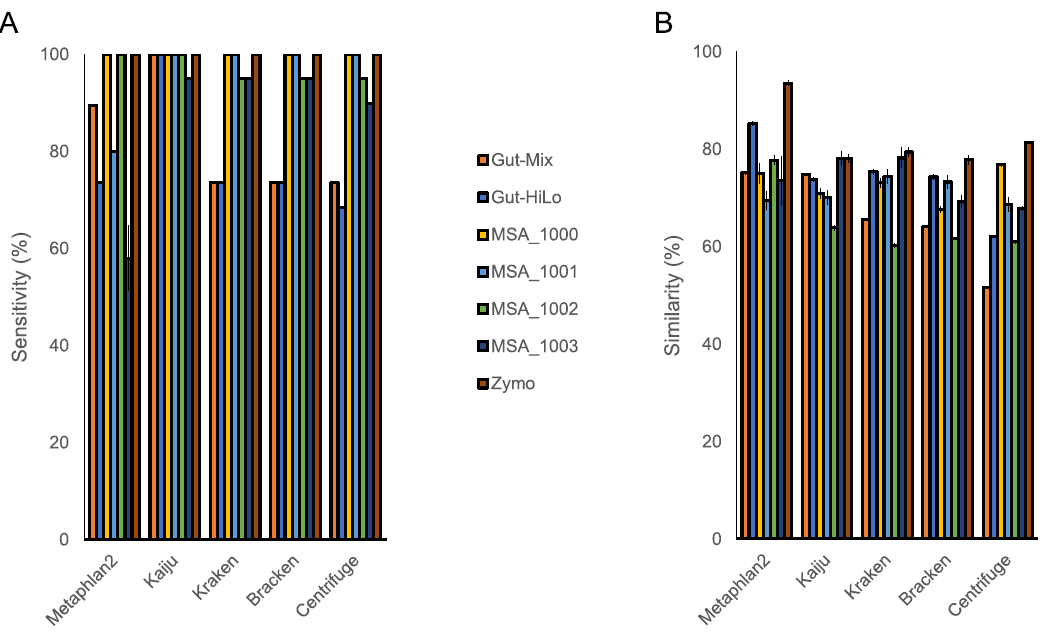
图3. 使用不同的参考试剂对生物信息学工具性能进行基准测试时,流程灵敏度(A)和相似性(B)的变化。Gut-HiLo = NIBSC Gut-HiLo-RR。Gut-Mix = NIBSC Gut-Mix-RR。MSA_1000 = ATCC MSA-1000。MSA_1001 = ATCC MSA-1001。MSA_1002 = ATCC MSA-1002。MSA_1003 = ATCC MSA-1003。Zymo = ZymoBIOMICS微生物群落标准
4. 生物分辨率影响流程性能
菌种的分类水平是生物信息学工具的重要特性,用户可利用它来研究菌种水平的异质性。为了测试生物信息学工具分辨低于种水平的能力,NIBSC-RRs包括了长双歧杆菌的两个亚种:B. longum ssp. longum and B. longum ssp. Infantis。在现有测序深度上,尚无流程可以准确地解析这两个不同的亚种,这表明使用浅度随机宏基因组学和生物信息学工具无法实现菌种或亚种的分离。
通常,根据研究假设和测序方法,微生物组研究报告有不同的分类级别。作者利用NIBSC-RR研究了变化的生物分类水平如何影响生物信息学工具的性能。使用Gut-Mix-RR时,四量度报告系统在属水平上的工具性能都得到了显着改善(图1)。五个工具中,有四个在属级别上具有100%的敏感性,有四个FPRA <1%,这对多样性和相似性都有影响,其中Kraken、Bracken和Kaiju有最高的相似度。MetaPhlAn2是唯一在较高的分类学分析中性能不会显著改善的生物信息学工具,但仍能对Gut-Mix-RR多样性进行最佳估计(图1)。对于Gut-HiLo-RR,分类水平对测量性能的影响较小,从种水平变为属水平分类,仅显著提高了灵敏度(图1)。在这两种参考试剂中,在较高分类标准下进行分类时工具性能的提高主要归因于先前分配给正确属的不正确种、现在被归类为正确属的读数。
5. 利用NIBSC RRs评估16S rRNA测序
16S rRNA扩增子测序是微生物组分类的最常用方法之一,并且是在大型队列研究中分析大规模微生物组变化的有效工具。考虑到它的广泛使用,作者测试了NIBSC RR和四步报告系统是否可以有效地对不同16S rRNA测序步骤进行基准测试,并探究它们可能会引入的偏差。通常,在不同的微生物组研究中使用不同的引物会扩增16S rRNA基因的不同区域。作者使用Gut-RRs,利用靶向V3-V4和V4区域的引物对SD-Bact-0341-bS-17 / SD-Bact-0785-aA-21、靶向V4区域的引物对515F(Parada)/ 806R(Apprill)测试了V3-V4和V4区域的性能差异,并利用QIIME2平台测试了两个最常用的分析扩增子数据流程性能的DADA2和Deblur。结果表明两种引物组均具有<0.01%FPRA的高度特异性;但515F(Parada)/ 806R(Apprill)引物相较于SD-Bact-0341-bS-17 / SD-Bact-0785-aA-21引物,敏感性、相似性、多样性均显著提高,这支持该引物组在所有微生物组研究中的广泛使用。使用两种参考试剂比较515F(Parada)/ 806R(Apprill)生成的DADA2和Deblur通道的数据,结果表明这些通道的性能相当。两条流程均以FPRA <0.01%检测到了15/16菌属,且两条流程给出的多样性估计值均与对Gut-Mix-RR和Gut-HiLo-RR观察到的属测量值一致。唯一不同的报告值是相似性,Deblur比DADA2与Gut-Mix和Gut-HiLo的实际成分具有更高的相似性。可用于扩增子测序的另一种方法是记录扩增子序列变体(ASV)的数量。Deblur始终估计两种试剂的ASV计数均为24,高估了20%;DADA2估计Gut-Mix-RR为26个ASV、Gut-HiLo-RR为27个ASV,分别高估了30%和35%。ASV对生物多样性的估计可能会因16S rRNA区域内基因组变异而增多。在这20个菌株中,共有53种可能的16S rRNA序列,这强调了使用ASV来衡量菌种多样性的问题,并支持先前有关使用DADA2导致高估真实菌株多样性的研究。
技术的飞速发展导致仅在过去十年中就出现了多种微生物组方法,评估不同方法之间的准确性是方法学的关键,这对于确保研究之间的可比性至关重要。在属水平上使用随机宏基因组测序和16S rRNA基因测序的四个度量报告系统进行计算,作者调查了是否可以准确比较这两种不同测序策略和相关生物信息学工具的数据(图1)。大致上,关于16S rRNA基因测序的四个报告结果与通过随机宏基因组测序获得的结果相似。在所有生物信息学工具中观察到了差异,但是这些差异是特定于工具/流程的,而不是所使用的库特定的偏差。例如,16S rRNA测序流程比随机宏基因组测序的MetaPhlAn2更敏感,却不如Kaiju敏感。同样,16S rRNA基因测序通道的FPRA值比Centrifuge、Kaiju低,但与MetaPhlAn2相同。这与既往研究一致,即浅度随机宏基因组测序测序和16S rRNA基因测序的结果在属水平上是可比的,生物信息学流程对本研究评估的四种报告值有关键影响。
结论:
作者开发了两种参考试剂(标准菌群DNA)和一个报告系统,可以帮助标准化微生物组领域的研究。对这些参考试剂的测试表明,它们可以准确评估生物信息学通道差异,并揭示了一系列shotgun测序生物分类学分析器之间的变异性。就使用此类试剂时用户应达到的共识阈值达成共识,可以防止错误报告数据,并可以协调该领域的研究。重要的是,参考试剂适合同时适用于16S rRNA测序和shotgun测序,并且它们的使用可比较两种不同方法的偏倚。同时使用通用参考试剂后观察到的通道性能差异,表明将来很有可能需要特定的参考试剂以确保正确地基准化。
后记:
微生物组学研究方法的标准化对于整个微生物组至关重要,也是推动微生物组学研究的关键步骤。本研究的作者开发出两种NIBSC DNA参考试剂及一套报告系统,简便通用,且能够比较不同的分析方法,这确保了参考试剂和报告系统能与既往研究方法兼容并进,实现了微生物组学研究方法的连续性和创新性。这将对肠道微生物组及皮肤、肺、口腔等的微生物组研究带来新的启发。
参考
Gregory C. A. Amos, Alastair Logan, Saba Anwar, Martin Fritzsche, Ryan Mate, Thomas Bleazard & Sjoerd Rijpkema. (2020). Developing standards for the microbiome field. Microbiome 8, 98, doi: https://doi.org/10.1186/s40168-020-00856-3
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读