昨天到脚本虽然顺利运行,但是路径太麻烦,没有检测输出文件是否qc成功。
下面是师兄脚本里的语句。


PE模式,HiSeq PE测序:
$ java -jar /path/Trimmomatic/trimmomatic-0.36.jar PE -phred33 -trimlog logfile reads_1.fq.gz reads_2.fq.gz out.read_1.fq.gz out.trim.read_1.fq.gz out.read_2.fq.gz out.trim.read_2.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
SE模式,HiSeq SE测序:
$ java -jar /path/Trimmomatic/trimmomatic-0.36.jar SE -phred33 -trimlog se.logfile raw_data/untreated.fq out.untreated.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
在SE模式中,是不需要指定文件来存放被过滤掉的read信息的,后面直接就接Trimmer信息!这是需要注意到的一个地方。
ILLUMINACLIP,接头序列切除参数。LLUMINACLIP:TruSeq3-PE.fa:2:30:10(省掉了路径)意思分别是:TruSeq3-PE.fa是接头序列,2是比对时接头序列时所允许的最大错配数;30指的是要求PE的两条read同时和PE的adapter序列比对,匹配度加起来超30%,那么就认为这对PE的read含有adapter,并在对应的位置需要进行切除【注】。10和前面的30不同,它指的是,我就什么也不管,反正只要这条read的某部分和adpater序列有超过10%的匹配率,那么就代表含有adapter了,需要进行去除;
链接:https://www.jianshu.com/p/36891a89ed6e
Running Trimmomatic
Paired End Mode:
java -jar
or
java -classpath
Single End Mode:
java -jar
or
java -classpath
If no quality score is specified, phred-64 is the default. This will be changed to an 'autodetected' quality score in a future version.
Specifying a trimlog file creates a log of all read trimmings, indicating the following details:
the read name
the surviving sequence length
the location of the first surviving base, aka. the amount trimmed from the start
the location of the last surviving base in the original read
the amount trimmed from the end
Multiple steps can be specified as required, by using additional arguments at the end.
Most steps take one or more settings, delimited by ':' (a colon)
Step options:
ILLUMINACLIP::::
fastaWithAdaptersEtc: specifies the path to a fasta file containing all the adapters, PCR sequences etc. The naming of the various sequences within this file determines how they are used. See below.
seedMismatches: specifies the maximum mismatch count which will still allow a full match to be performed
palindromeClipThreshold: specifies how accurate the match between the two 'adapter ligated' reads must be for PE palindrome read alignment.
simpleClipThreshold: specifies how accurate the match between any adapter etc. sequence must be against a read.
SLIDINGWINDOW::
windowSize: specifies the number of bases to average across
requiredQuality: specifies the average quality required.
LEADING:
quality: Specifies the minimum quality required to keep a base.
TRAILING:
quality: Specifies the minimum quality required to keep a base.
CROP:
length: The number of bases to keep, from the start of the read.
HEADCROP:
length: The number of bases to remove from the start of the read.
MINLEN:
length: Specifies the minimum length of reads to be kept.
Trimming Order
Trimming occurs in the order which the steps are specified on the command line. It is recommended in most cases that adapter clipping, if required, is done as early as possible.
安装fastqc
$ unzip fastqc_v0.11.5.zip
$cdFastQC
$ chmod 755 fastqc
运行$ /path_to_fastqc/FastQC/fastqc untreated.fq -o fastqc_out_dir/
命令比较简单,这里 唯一值得注意的地方就是 -o 参数用于指定FastQC报告的输出目录,这个目录需要事先创建好,如果不指定特定的目录,那么FastQC的结果会默认输出到文件untreated.fq的同一个目录下。它输出结果只有两个,一个html和一个.zip压缩包。
我们可以直接通过浏览器打开html,就可以看到FastQC给出的所有结果,zip压缩包解压后,从中我们也可以在对应的目录下找到所有的QC图表和Summary数据。
qc前的fastqc图

qc后的序列fastqc图
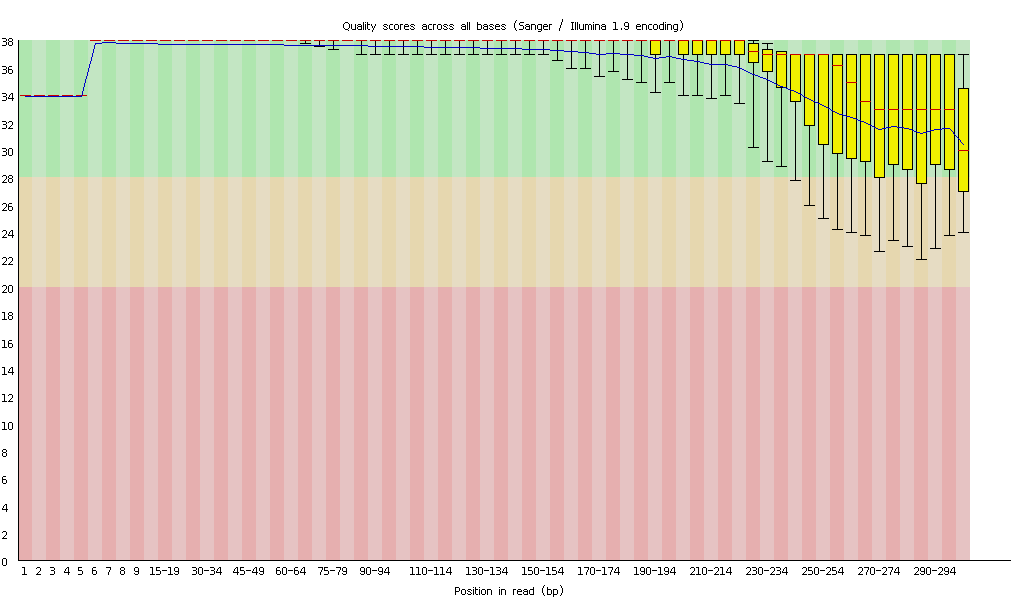
疑问:双端测序的两个文件都需要考虑到吗?
Single-end、Paired-end主要区别在测序文库的构建方法上。
1、单端测序(Single-end)首先将DNA样本进行片段化处理形成200-500p的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flowcell上生成DNA簇,上机测序单端读取序列。
2、Paired-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paried-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序。