AlignNemo:一种融合同源和拓扑的局域网络对齐方法
摘要
局部网络比对是蛋白质-蛋白质相互作用网络分析的重要组成部分,可能导致进化相关复合体的识别。我们提出了一种新的算法AlignNemo,它在给定两个有机体的网络的情况下,揭示了与生物功能和相互作用拓扑相关的蛋白质子网络。所发现的保守子网络具有一般的拓扑结构,不需要对应于特定的相互作用模式,因此它们更接近于文献中提出的功能复合体模型。该算法能够通过扩展过程来处理稀疏的交互数据,该扩展过程在每一步都会探索超出与当前解决方案直接交互的蛋白质之外的网络的局部拓扑。为了评估AlignNemo的性能,我们使用统计方法和生物学知识进行了一系列基准测试。基于蛋白质复合体的参考数据集,AlignNemo在准确率和召回率方面均优于其他方法。通过将语义相似度的概念应用于基因本体论词汇表,我们证明了我们的解决方案在生物学上是合理的。有关AlignNemo的二进制文件以及有关算法和实验的补充细节,请访问:sourceforge.net/p/alignnemo。
绪论
在过去的几十年里,研究人员一直关注进化在基因组水平上的影响,即如何通过分析基因组序列来重构进化。最近,关于蛋白质-蛋白质相互作用的高通量数据的可获得性使人们能够通过比较不同物种的蛋白质相互作用图(也称为相互作用图)来观察进化变化[1-3]。该领域的目标包括识别物种间相互作用的保守模式,以及识别新的正交关系[4]。在这种情况下,已经开发了几种用于比较蛋白质-蛋白质相互作用(PPI)网络的算法,通常被称为网络比对算法。
网络比对问题有两个主要实例:全局比对通过搜索来自不同物种的一整套蛋白质和蛋白质相互作用的单一综合图谱来回答进化问题;局部比对搜索细胞机械的进化保守的构件,而忽略网络之间的总体相似性。图论中的形式主义提供了解决这两个问题的最佳框架。在这种形式下,PPI网络被表示为图(G),图(G)的节点(V)是蛋白质,边(E)是它们之间的交互。蛋白质网络对齐问题被描述为一个图对齐问题,即在两个(成对的)或多个(多个)的图之间寻找相同或相似的子图。形式上:给定两个输入图G1~fV1,E1g和G2~FV2,E2g,G1和G2的对齐问题可以表示为寻找G1中的节点和G2中的节点之间的映射M(M:V?1?V?2,其中V?1(V1,V?2(V2))使定义在节点和边上的关联相似性函数最大化。对于全局对齐,M是网络的整组节点之间的映射。相比之下,对于局部对齐,M被定义为最相似的节点子集之间的映射集。本文针对PPI网络的局部比对问题,提出了一种在两个PPI网络中提取保守蛋白质复合体的方法。
蛋白质复合体在这里被定义为执行相似功能或参与相同生物过程的一组蛋白质。现有的检测蛋白质复合物的方法通常是基于这样的观察,即复合物对应于高度相互作用的蛋白质集合,因此他们在PPI网络中寻找稠密的子图。例如,两个版本的NetworkBLAST[5,6]都基于这样的假设,它们是从最初专注于保守路径的PathBLAST[7]演变而来的。在我们提出的方法中,我们寻找相对密集的节点组,即它们之间的交互比与网络的其余部分具有更多的交互,从而对复杂的拓扑施加不那么严格的约束。事实上,虽然拓扑是信息性,它通常被证明是不完整的,反映了对蛋白质的不统一知识[8,9]。几个假阴性的存在导致稀疏的图,甚至更稀疏的物种之间的保守相互作用集,使得只寻找稠密的子图的方法无法检测到保守的复合体。
有几种方法,如NetworkBLAST,依赖于在称为比对图的结构上搜索保守的复合体。比对图具有对应于成对的同源蛋白的节点和保守相互作用的边。为了处理丢失的信息,NetworkBLAST以及类似的方法引入了不那么严格的比对图定义,通过允许在原始PPI网络中的相应的直系蛋白质对距离小于或等于k(对于NetworkBLAST k~2)的情况下节点连接。然而,以这种方式,即使对于较小的k值,也可能向对齐图添加几个不可靠的链路,从而导致不正确的解。
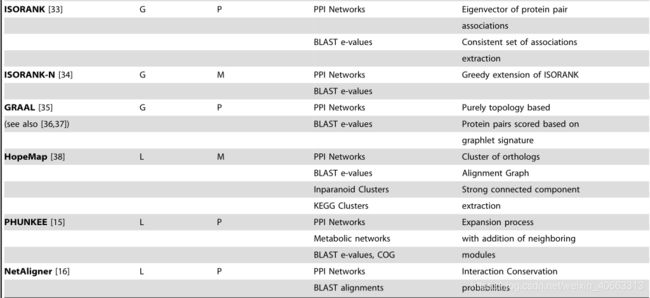
表1.关于网络调整工具的概要。
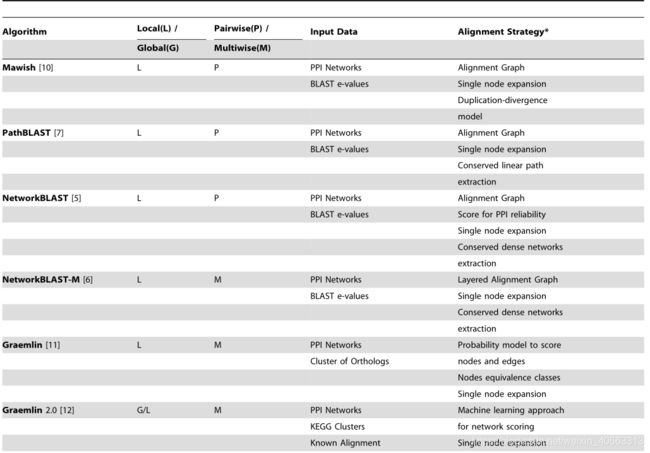
Mawish方法[10]将网络对齐作为一个最大权导出子图问题,结合进化模型来评估拓扑相似性。虽然有效,但正如我们在实验中观察到的那样,这个模型可能过于严格,无法识别较小的保守结构,并且无法恢复较大的复合物。
其他算法,如Graemlin[11]及其新版本Graemlin 2.0[12],通过允许搜索更一般的拓扑来推广以前的方法。这些方法提高了检测有意义的比对的能力,除了同源信息外,还使用了来自Inparanoid的蛋白质之间的同源关系[13]、KEGG途径注释[14]和已知的比对。然而,这些方法没有充分利用拓扑信息,因为局部比对步骤只检查每个节点的直接邻域,以贪婪的方式迭代地分组最佳邻域。
Punkee[15]在网络环境中考虑局部保守的子网络方面向前迈进了一步:在选择假设的同源序列集合之后,该方法同时探索所有相邻的蛋白质以寻找高度保守的相互作用集合。但是,所有交互都具有相同的可靠性,并且由Phunkee定义的网络环境不会超出直接交互的范围。最后,与这项工作的发展同时,一种新的方法,NetAligner[16],设计了一个蛋白质组比对的算法框架。NetAligner引入了一种策略来识别进化保守的相互作用,这依赖于相互作用的蛋白质以比偶然预期的更接近的速度进化的原理。
虽然对可用于全局和本地网络对齐的算法的详细描述超出了本白皮书的范围,但表1中提供了有关可用工具的更广泛的概要。
我们在这里介绍一种方法,AlignNemo(对齐网络模块),它通过为本地网络对齐提供一个通用而有效的框架来解决上述问题。AlignNemo通过不同的步骤进行操作,如图1所示。首先,它从输入网络构建加权对齐图。节点代表假定的同源蛋白质对,并与Inparanoid中一样进行评分,反映了绘制蛋白质对的置信度。相比之下,用一种新的方法对边进行加权,该方法考虑了输入网络中的本地连通性(参见方法)。然后,我们从对齐图中提取所有给定大小的连通子图,并根据节点和边上的权重对它们进行排序。排名靠前的完全连通子图将被用作对齐解的种子。最后,我们通过在每个步骤添加多个子图,以迭代的方式扩展每个种子。这使我们能够探索解决方案的网络环境,使其超越其直接邻居。在方法一节中提供了算法的正式描述。
该方法的主要贡献是:1)一种新的比对图边评分策略,它通过输入PPI网络在两个给定节点之间的路径集合来分析它们的结构,并估计它们的可靠性和局部重要性;2)一种新的迭代扩展过程,它从种子开始,在除直接交互之外的每一步探索比对图的局部拓扑。这种组合提供了一种新的方法来解释拓扑和同源性,并被证明在检测大量不同的蛋白质复合物时被证明是有效的,而不依赖于它们的大小或连接度。
在下一节中,我们展示了酿酒酵母、黑腹葡萄球菌和智人PPI网络的比对结果作为原理的证明。我们证明了我们的比对比其他方法具有更好的拓扑和生物学质量。结果的质量通过不同的方法进行评估:首先,我们通过准确率和召回率的衡量,展示了AlignNemo恢复已知蛋白质复合体的能力;然后,我们将语义相似度的概念应用于Gene Ontology词汇,证明了我们的解决方案在生物学上是合理的;最后,我们表明,即使在该方法施加的限制较少的情况下,所提取的模块也保持了高连通性。
详细讨论了具有代表性的复合体,并提供了与诸如NetworkBLAST、MAWISH和NetAligner等局部比对工具的比较,作为该软件可用和当前维护的仅有的复合体。我们选择NetworkBLAST和MaWish进行主要分析,因为它们可用于用户定义的输入数据,而我们分别比较了AlignNemo和NetAligner,因为我们根据其自己的数据和交互概率运行后者。Http://www.bioinformatics.org/alignnemo.上提供了AlignNemo以及本文中使用的适当文档和数据集
结果和讨论
在这一部分中,我们评估AlignNemo,NetworkBLAST和Maish在黑腹果蝇(果蝇),酿酒酵母(面包师酵母)和智人(人类)的公共可用数据集上的性能。我们在相同的数据集上运行这些方法,每个算法产生一组可能重叠的解决方案或模块。模块M是包含来自两个输入网络的一组蛋白质对的比对图的子图。我们将M中来自网络G1和G2的蛋白质集分别称为MG1和MG2。
从同调和拓扑的角度对每种方法的解进行了评估和比较。首先,我们证明了AlignNemo能够以较高的精确度和召回率重现已知的蛋白质复合体。然后,我们将语义相似度的概念应用于基因本体论词汇,证明了来自不同物种的蛋白质之间的联系在生物学上是合理的。最后,我们偶然地证明了我们的解比预期的更紧密地联系在一起。在本节结束时,我们将重点介绍几个具体的案例,以突出每种方法的缺点和优点。
输入数据
黑腹葡萄球菌和酿酒酵母的蛋白质-蛋白质相互作用来源于相互作用蛋白质数据库(DIP于2011年10月27日更新)[17]。在果蝇中包括7,548个蛋白质和22969个相互作用;在酵母中包括5,053个蛋白质和22254个相互作用。用Inparanoid[13]从这两个网络中筛选出10045对可能的同源蛋白,其中酵母蛋白1,878个,果蝇蛋白1,511个。智人PPI网络来自嬉皮士数据库[18];它包括来自17个不同来源的12113个蛋白质和78559个加权相互作用。从Gerstein实验室获得了一组假定的人和苍蝇的同源蛋白对[19]。
这些数据集集成了多个来源,并包括来自不同方法的交互,包括高通量和小规模实验。为了说明这种多样性,我们为每条边分配了可靠性分数。对于来自DIP(果蝇和酵母)的两个网络,我们采用了[20]中定义的最大似然估计程序来评估通过相同的实验程序确定的蛋白质相互作用的可靠性。该方法基于不同时间点的基因表达谱的相关性是评价PPI可靠性的良好特征:相互作用的蛋白质通常表现出很高的相关值。在应用这一方法时,我们将未知相互作用的随机蛋白质对视为真正的非相互作用蛋白质,将由小规模实验确定的相互作用视为真正相互作用的蛋白质,从这两个集合估计各自的相关系数分布。对于酵母蛋白,我们使用了SGD数据库[21]中报告的一组表达谱,并为DIP中描述的每种实验方法和它们的组合分配了置信度分数。苍蝇相互作用的分数是基于给定的实验方法在不同生物体中同样有效的假设来计算的,因此基于酵母数据的置信度分数被转移到苍蝇相互作用中。人类蛋白质相互作用网络的可靠性分数可以通过网络服务器HIPIE获得。
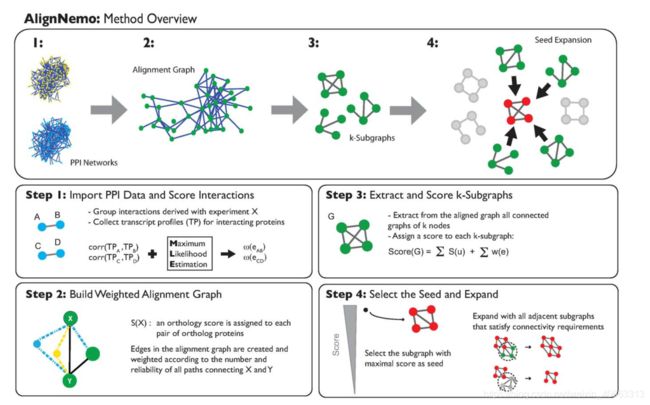
图1.AlignNemo概述。给定两个输入PPI网络(1),构建对齐图,并为其节点和边(2)分配分数。然后,从对齐图(3)中提取种子,即具有大量高得分链接和节点的小的子图,并且通过添加通过可靠的链路(4)相对良好地连接到每个种子的小的子图,以贪婪的方式扩展每个种子。Doi:10.1371/Joural.pone.0038107.g001
已知络合物的检测
我们通过评估每种方法找到的模块与已知配合物的一致性来评估结果的质量。给定一个模块和一个已知的复合体,我们计算了信息检索中两个广泛使用的度量:精确度(P)和召回率(R)。精确度定义为模块中也存在于复合体中的蛋白质的百分比;召回率定义为复合体中也存在于模块中的蛋白质的百分比。为了将这些度量集成到单个分数中,我们计算F1分数函数,该函数被定义为准确率和召回率的调和平均值。从形式上讲,这些措施的定义如下:

其中Tp是真阳性的数量,即在溶液中发现的也在复合物中的蛋白质的数量。类似地,fp和fn是假阳性和假阴性的数量。F1-得分范围在[0,1]区间内,1对应于完全一致。在我们的分析中,我们从给定的算法中匹配物种Gito的所有模块MGi中的每个已知复合体,并选择F1得分最高的模块作为最佳匹配的模块。
为了评估酿酒酵母和黑腹葡萄球菌的比对结果,我们参考了CYC2008中的复合物[22],这是一个来自小规模实验和文献挖掘的408个酵母蛋白复合物的综合目录。为了对黑腹毛虫和智人进行比对,我们参考了CORUM[23]中的复合体,这是一个包含1682个人类蛋白质复合体的数据集。我们观察到28%的CYC2008和CORUM复合物仅由2或3个蛋白质组成(CYC2008为132个蛋白质,CORUM为474个蛋白质)。这可能是有问题的,因为对于这样小的综合体来说,统计指标往往很难解释。为此,我们将我们的分析限制在至少含有4个蛋白质的复合物上,但同时我们验证了每种方法回收小复合物(2-3个蛋白质)的能力。我们考虑了一个小的复合体,如果它的至少2个蛋白质与一个比对溶液重叠,不包括超过20个节点的溶液,就可以恢复。在表2中,我们总结了四种算法的性能。在表格中,我们列出了每个算法找到的模块的数量,其中还列出了高质量模块的数量,即那些与F1得分大于0.3的已知复合体匹配的模块。AlignNemo、Mawish和NetworkBLAST获得的F1分数的总体分布通过各自的内核密度分布进行估计,如图2(A-B)所示。在图2(A-B)中,我们还分别报告了每种方法在查准率和召回率方面的性能。NetworkBLAST和AlignNemo在酵母-苍蝇比对上都表现得更好,后者的准确率和召回率总体上都更高。马维希找到的小解一般都有很高的精度。
F1分数的完整列表,以及精确度和召回率的衡量标准,都可以作为补充材料(表S1)。对于每一场比赛,我们还报告了经多次检验校正后的Fisher精确检验得出的p值。AlignNemo在恢复已知复合体方面明显优于其他方法,显示出高质量模块的最高百分比。应该注意的是,虽然Mawish对于飞人对齐的性能类似地很好,但是由该方法产生的大多数模块都具有小尺寸,具体地说,90%的模块只由2个节点组成。
物种间的蛋白质定位
在上一节中,我们展示了AlignNemo能够概括已知的蛋白质复合物,并且检测到的保守子网络通常反映了每个单一物种中的已知生物学。另一方面,不同物种间蛋白质图谱的质量有待进一步评价。我们根据功能相似性来评估所发现的映射的生物学相关性,即我们确定来自两个生物的匹配蛋白质在功能上相关的程度。
这种分析需要使用编码到本体论中的先验生物学知识。我们选择基因本体论(GO)框架及其注释来确定来自不同物种的两个蛋白质之间的功能相似性,使用语义相似性的概念[24]。在我们的分析中,我们使用GO中的生物过程(BP)和分子功能(MF)本体的注释集计算了每个解决方案的语义相似度。我们在这里仅报告BP的结果,因为这个本体更紧密地反映了蛋白质复合物作为参与特定过程的亚细胞单元的想法。表S2中报告了完整的结果。
给定两个蛋白质p1和p2及其GO(P1)和GO(P2)组,Resnik相似性度量[25]用于用GOI[GO(P1)和GOJ[GO(P2)]对每对(GOI,GOJ)进行评分。根据Resnik度量[26],p1和p2的语义相似度被定义为围棋(P1)和围棋(P2)中的每个围棋术语的最佳匹配得分的平均值。使用工具FastSemSim[27]计算语义相似度。
我们总共测试了AlignNemo的356个解决方案,其中85%的解决方案含有5到15个蛋白质,最大的93个蛋白质;NetworkBLAST的362个解决方案,每个包括5到15个蛋白质,后者是该方法施加的限制;以及Mawish的260个解决方案,每个包括2到6个蛋白质。鉴于检测到的子网的大小存在显著差异,我们在图2(C-D)中分别针对小复合物(v7蛋白质)和大复合物(§7蛋白质)显示了这三种方法获得的结果。
对两种蛋白质网络比对的结果表明,三种算法在语义相似性方面的性能相似,而对于H.sapiens-D.Blackogaster蛋白质的比对性能更好。
表2.AlignNemo、MaWish、NetworkBLAST和NetAligner的比较。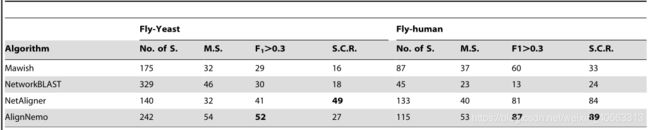
不是的。OF S:解数;M.S:匹配解;S.C.R:回收的小复合体。每种算法找到的解的数量(No.。(S.)。分别列在第2栏和第5栏中,分别用于酵母-苍蝇和苍蝇-人的比对。与至少一个已知复合物匹配的溶液的数量在列3和列6(M.S.-匹配解决方案)中针对每个比对进行报告。尺寸§4的复合物的高质量匹配数汇总在第4栏和第7栏(F1w0:3),而回收的小复合物(2-3个蛋白质)的数量在第5栏和第8栏(S.C.R.-回收的小复合物)。Doi:10.1371/Joural.pone.0038107.t002
守恒模的拓扑
在这里,我们分析了所得解的拓扑结构。正如在引言中所讨论的,蛋白质复合物通常由紧密相互作用的蛋白质组成。然而,最近关于PPI网络中复合物的模块性和组织的研究结果表明,它们往往由一个紧密连接的核心和一个连接强度较低的一组蛋白质定义的附着组成。后者通常存在于多个复合物中,并且允许潜在功能的多样化[28]。
根据这个模型,AlignNemo寻找相对紧密连接的蛋白质,即它们之间的相互作用比网络的其余部分更多的蛋白质,而不是对候选解决方案的拓扑施加严格和固定的限制。
我们现在想测试这一策略是否会危及我们检测密集连接核心的能力,包括我们的解决方案中不太可能是真正的蛋白质复合物的稀疏子网络。为此,我们为每个PPI网络生成1000个随机网络,保持它们的节点度分布,然后我们评估每个模块的连通性,即边数,在原始PPI网络和随机集合中。因此,对于每个物种和每个解决方案,我们估计其连通性的背景分布。我们使用Z分数来量化实际网络中观察到的连通性Ci与这样的背景分布的偏差:
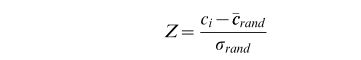
其中c crand值是随机集合中这组蛋白质的平均连接性,并显示其标准偏差。
首先,我们分别测试由每个解决方案定义的两组蛋白质,每个物种一个,然后,我们将获得的两个之间的最大Z分数与每个解决方案相关联。通过这种方式,我们也解释了一个物种中相对连接较差的蛋白质,而另一个物种中相应的同源基因是密集相互作用的。从该背景分布中经验地为每个模块导出p值,并且它由在所有可能的网络上导致测试模块的Z分数更大或相等的随机网络的数量给出。有趣的是,我们发现95%的解决方案,包括人-蝇和酵母-蝇的比对,都显示出比随机网络中观察到的显著更高的连接性。
总而言之,AlignNemo在正确检测单个物种内的蛋白质复合体方面优于Mawish和NetworkBLAST,因为它们之间存在相互作用和同源关系。此外,不同物种之间的蛋白质映射在生物学上是合理的,正如同一模块中蛋白质之间的平均语义相似性所证明的那样。最后,尽管AlignNemo没有对模块拓扑施加严格的限制,探索蛋白质复合体的不太强连接的组件,但提取的子网的连接密度比预期的要高。


图2.AlignNemo、NetworkBLAST和Maish的比较。根据在S:Cerevisiae(CYC2008)和H:Sapiens(Corum)中恢复已知蛋白质复合物的情况,对这三种算法进行了评估。与已知复合体匹配的解决方案通过精确度、召回率和F1得分进行评分。获得的每种方法的分数分布被绘制在图(A)中(酵母-苍蝇比对)和图(B)中,用于人-蝇比对。面板(C)和(D)显示了来自不同物种的蛋白质之间的平均语义相似性,这些蛋白质由每种溶液绘制而成。每个解由一个半径与解的大小成正比的圆表示。每种方法的解决方案的大小差别很大,因此分别显示了小的(7个节点)和大的(§7个节点)解决方案。*百分比是指通过至少一种方法匹配的复合体集合。Doi:10.1371/Joural.pone.0038107.g002
与NetAligner的比较
NetAligner依靠一种新的算法方法,基于来自不同物种的蛋白质之间的蛋白质序列相似性,计算与保守相互作用相关的概率。给定两对假定的同源基因,NetAligner通过考虑两对同源基因之间进化距离的差异来评估它们共享保守相互作用的可能性。我们在不同的配置和输入数据下测试了NetAligner,包括工具提供的原始蛋白质组和同源性。根据我们的分析,NetAligner在使用预测可能守恒的相互作用设置以及其参考论文[16]中建议的参数时,性能最佳。NetAligner在其自己的数据集上提取更大、更可靠的比对集合。因此,我们决定比较AlignNemo和NetAligner各自在自己的数据集上运行。
当溶液与参考复合物(CYC2008和CORUM)匹配时,两种方法的执行情况相似(参见图3和表2)。AlignNemo再次显示S:Cerevisiae-D:Blackogaster排列的整体性能更好。在H:SAPINS-D:Blackogaster比对中,NetAligner找到了一组得分更高的小解决方案,但同时由一个包含463个节点的非常大的解决方案产生了几个匹配,从而导致高召回值,尽管精确度接近于零(图3)。
守恒复合体
在这一部分中,我们特别关注CYC2008和CORUM的几个复合体,以更好地剖析不同方法的性能。这里讨论的案例包括一个小的复合体,Arp2/3,以及两个相对较大的复合体,TFIID(通用转录因子)和20S蛋白酶体,它们具有不同水平的连接性。在表3中,我们报告了这些复合物的蛋白质在H:Sapiens和D:Blackogaster网络比对中被AlignNemo、NetworkBLAST和Mawish之间的至少一个正确结合和恢复的蛋白质。对于转录因子TFIID和Arp2/3复合物,AlignNemo根据F1得分和语义相似性都表现得更好。在检测20S蛋白酶体时,AlignNemo和NetworkBLAST的酵母-苍蝇比对召回率相当,但AlignNemo的准确率更高。此外,AlignNemo在人与苍蝇对齐方面显示出卓越的性能。使用GOTermFinder[29]计算了我们解决方案的显著丰富的GO类别,并在表S3中进行了报告。在这两个比对中,AlignNemo的跨物种语义相似性较高,表明生物质量有所改善,具体内容如下所述。
转录因子TFIID复合物
RNA聚合酶(I、II和III)催化核基因的转录,依赖普通转录因子识别目标启动子,特别是RNA聚合酶II依赖TFIID复合物启动转录。通用转录因子TFIID主要由TATA盒结合蛋白(TBP)和一组跨物种高度保守的TBP相关因子(TAFII)或亚基组成[30]。
AlignNemo在发现这一复合体方面优于现有的方法:它在19个节点的溶液中发现了9种TFIID蛋白质;它正确地将人类蛋白质映射成与两个生物体中同一亚单位相对应的飞行蛋白质(见表3)。Mawish的特点是只有2个节点的解决方案,也包括在我们的比对中,而NetworkBLAST返回的10个节点的解决方案与属于TFIID复合物的4个蛋白质对相匹配。
由于该复合体的高度连接性,AlignNemo和NetworkBLAST解决方案超出了CORUM中定义的TFIID复合体的范围。为了进一步验证这些溶液的质量,我们对其中的所有蛋白质进行了GO项的富集测试。我们发现在AlignNemo‘s溶液中17个飞行蛋白中的16个和19个人蛋白中的18个具有相同的GO条件,包括RNA聚合酶II启动子(PFLY~1:21E{23,phuman~8:16E{18)的转录。相比之下,NetworkBLAST的解决方案只报告了两个网络中具有共同和特定生物学作用的10个蛋白质中的4个(参见表S3)。
Arp2/3复合体由7个单位组成,在肌动蛋白细胞骨架的调控中起重要作用。它是肌动蛋白细胞骨架的主要组成部分,在大多数含有肌动蛋白细胞骨架的真核细胞中都有发现[31]。
有趣的是,在最初的PPI网络中,这些蛋白质之间的连接水平差异很大,从在人类中发现的17种相互作用到在D:Blackogaster中没有发现的相互作用。不完整的信息使这一复杂的恢复变得特别困难。事实上,只有AlignNemo能够在H:Sapiens和D:Blackogaster中鉴定到这个保守的复合体,而NetworkBLAST和Maish都没有与之重叠的任何解决方案。表3列出了在AlignNemo溶液中发现的正确检测到的同源蛋白。所有4个都标注有肌动蛋白微丝聚合功能GO项的调节(PFLY~3:07E{08和Puman~1:24E{09)。这个案例很好地指出了考虑保守路径(而不仅仅是直接交互)来补充一个网络中缺失信息的重要性。

图3.AlignNemo和NetAligner的比较。这两种算法在恢复S:Cerevisiae(CYC2008)和H:Sapiens(Corum)中的已知蛋白质复合物方面进行了评估。与已知复合体匹配的解决方案通过精确度、召回率和F1得分进行评分。Doi:10.1371/Joural.pone.0038107.g003
表3.Arp 2/3、TFIID和20S蛋白酶体复合物的最佳匹配溶液的比较。
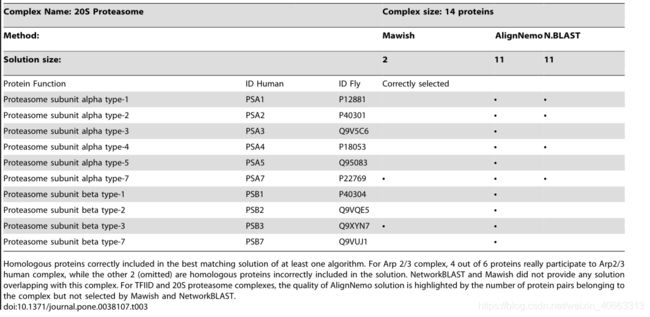
20S蛋白酶体复合体
20S蛋白酶体是存在于几种生物体中的一种大型蛋白质复合体,特别是在这里研究的所有三种生物体中。根据CYC2008和CORUM的研究,20S蛋白酶体在酵母中由14种蛋白质组成,在人类和苍蝇中由16种蛋白质组成。络合物的拓扑结构相对密集,相互作用可靠。
对于S:Cerevisiae-D:Black ogaster网络比对,三种方法的召回值都相当;至于精确度,NetworkBLAST因为在复合体之外发现了几个蛋白质,所以得到的值要低得多。另一方面,AlignNemo在识别H:Sapiens-D:Blackogaster网络比对中的20S蛋白酶体复合体方面优于其他方法(参见表3)。事实上,它正确地选择了人类20S蛋白酶体的11个蛋白质和苍蝇的12个蛋白质,而NetworkBLAST在两个网络中只发现了4个人和5个果蝇的蛋白酶体和2个蛋白酶体。
方法
AlignNemo旨在识别不同物种的PPI网络之间保守的蛋白质模块或复合物。搜索保守模块是在比对图上执行的,由三个主要步骤组成。
- 首先,根据输入网络构造对齐图。比对图中的每个节点对应于一对假定的同源蛋白,来自Inparanoid的分数被用来对每个节点进行加权。根据计分策略对对准图的每条边进行加权,该计分策略结合了关于在输入网络中连接其端点的路径的数量、可靠性和局部重要性的网络上下文信息。该策略通过辅助结构(联合图)来实现,该辅助结构对该方法的整体性能至关重要。
- 其次,从对齐图中提取所有连通的k-子图(这里是k~4),并根据结点和边的权重进行评分。排名靠前的全连通k-子图将被用作对齐解的种子。
- 第三,通过探索当前解的超出其直接邻居的局部邻域,以迭代的方式扩展每个种子。具体地说,我们定义了一个扩展过程,该过程在每个步骤添加所有通过可靠交互与当前解决方案(而不是网络的其余部分)连接更紧密的子图。
这一方法与最近关于网络中复合体的模块性和组织的研究结果一致,根据该发现,PPI网络中的复合体往往由核心部分和附件组成。核心被定义为一小群功能相似并具有高度相关的转录图谱的蛋白质。核心被连接程度较低的蛋白质所包围,这些蛋白质是限定的附着体,存在于多个复合体中,允许潜在功能的多样化[28]。这种多样化在我们的解决方案结构中得到了很好的反映。实际上,如前几节所示,我们确定了几个重叠的模块,而不是没有交集的独立子网。
对齐图
比对图GA~(VA,EA)是一个加权图,图中的节点代表成对的同源蛋白质和边守恒的相互作用。如前所述,对齐图的现有定义在两个节点之间设置边的方式上有所不同。大多数表示都利用了来自输入的有限数量的拓扑信息,因为它们几乎丢弃了几乎所有没有参与同源关联及其交互的节点。
我们的目标是构建一个尽可能多地考虑这两个网络结构的比对图。我们设计了一种新的比对图边评分策略,该策略综合了原始网络中存在的拓扑信息,即两个节点之间长度小于等于2的路径的数量、可靠性和重要性。通过引入一种称为并图的辅助结构,该策略得到了最好的描述和实现。比对图的构建和评分包括三个步骤:(I)将所有输入的网络数据合并到并图中,(Ii)对并图进行处理以生成原始的比对图,最后(Iii)对原始的比对图进行一些剪枝操作,以去除噪声,提高整体计算速度。
并集图
联合图的目的是在不丢失信息的情况下将所有输入数据合并到单个图中。给定两个加权网络G1~(V1,E1)和G2~(V2,E2),以及G1和G2的节点之间的一组同源关联H~(u,v),u[V1,v[V2fg],并图U(G1,G2,H)包含两种类型的节点:(I)表示由H列出的同源蛋白质对的复合节点,以及(Ii)表示不具有同源的两个输入网络的蛋白质的简单节点。包含在输入网络之一中的任何边通过在所有对应节点对之间添加边来表示在并图中,所述对应节点可以是简单的,也可以是复合的。正式地:
定义1.并图U(G1,G2,H)~(VU,Eu)是具有如下结构的图:

假设E1和E2的每个边e用可靠性分数w(E)标记,并且每个关联k[H用可靠性分数w(K)标记。则U(G1,G2,H)中的边(i,j)被分配由输入网络中的对应边的分数给出的分数w(i,j);唯一的例外是当i和j都在VC中时,即,它们是合成节点,并且在两个输入网络中都有对应的边,在这种情况下,w(i,j)是两个原始边的分数之和。
图4给出了一个联合图的结构示例。
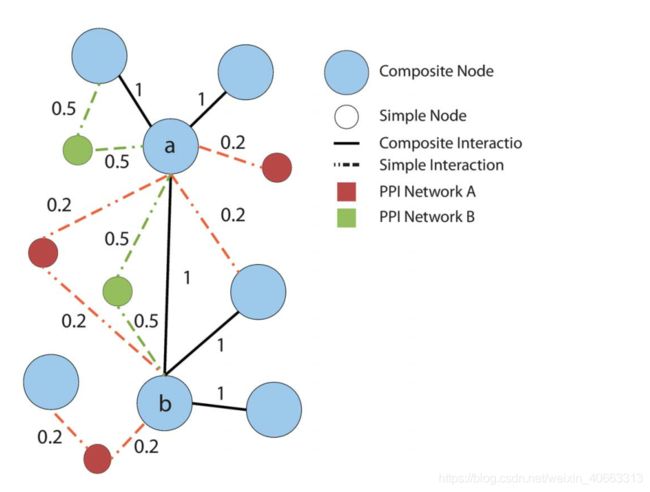
图4.联合图的示例。联合图既包括表示来自两个物种的同源蛋白对的复合节点(浅蓝色节点),也包括表示在另一个网络中没有同源的蛋白质的简单节点(红色和绿色节点)。类似地,复合交互(黑边)和仅存在于一个物种中的交互(红边和绿边)都出现在并集图中。
原始对齐图
对齐图GA~(VA,EA)可以看作是联合图的一个简化版本,在联合图中,如果两个结点之间至少有一条长度小于等于2的路径,则只保留合成结点,并由一条边连接两个结点。长度为2的路径的中间节点可以是简单的,也可以是复合的。对齐图定义中最重要的部分包括边评分策略,该策略通过考虑连接并图中满足特定标准的两个节点的所有路径来总结并图的局部拓扑。这一策略是基于这样的假设,即通过大量路径连接的同源蛋白很可能在功能上是相关的。因此,两个节点之间的每条路径都提供了它们相关性的附加证据。
在并图中考虑距离不大于2的节点对的选择似乎是合理的。一方面,仅考虑直接连接的节点对不适合于对进化中的远距离物种进行比对,并且对原始PPI网络中的缺失交互不具有健壮性。另一方面,我们的实验表明,在距离大于2的节点对之间添加边会显著增加对齐图的边数,而不会在结果质量方面提供任何好处。必须注意,联合图中长度为2的一些路径是虚假的,即它们不对应于输入网络中的路径。这样的路径在我们的分析中被忽略了。
长度为2的路径(以下称为间接路径)由于原始PPI网络中缺少交互而起主要作用。然而,并不是所有的间接路径都具有相同的意义。具体地说,间接途径可能通过高度或松散相互作用的蛋白质。如果节点在并图内高度交互,则两个节点通过它通信的概率很高。此外,组成不同路径的边可以具有不同的置信度分数,并且可以表示保守或非保守的交互。
为了将所有这些观察结果考虑在内,我们设计了一个基于贾卡德指数的新分数[32]。对齐图中的每条边EA~(a,b)基于连接a和b的长度为2的路径的数目进行评分。GA的两个节点a和b之间的边的最终分数由两项之和给出:直接贡献S1和间接贡献S2。直接贡献被评估为联合图(如果存在)中连接a和b的直接路径(a,b)的分数除以将a或b连接到联合图中的任何其他复合节点的所有直接路径的分数之和的比率。类似地,间接贡献被评估为联合图中连接a和b的长度为2的路径的分数除以将a或b连接到联合图中的任何其他合成节点的所有长度为2的路径的分数之和。形式上,我们将连接两个复合节点的路径集合定义为它们的扩展局部交互体,并导出如下分数:
定义2-扩展局部互动(ELI)评分。设w(a,b)表示并图中连接节点a和b的边的分数(w(a,b)~0,如果(a,b)=[ea),且w(Pab)~w(a,i1)z…zw(ik{1,b)是连接a和b的长度为k的路径的分数),则如果Ek(A)是连接a到距离为k的邻居的路径的集合,并且w(Ek(A))是与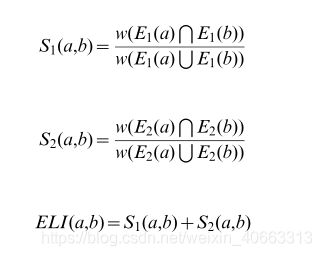
这种计分策略的力量依赖于它再次考虑对齐节点的局部邻域的能力:虽然像NetworkBLAST或Maish这样的方法允许间隙或失配来连接对齐图中距离为2的保守蛋白质,但我们考虑到连接保守蛋白质对的整个路径集及其可靠性。
图4中给出了一个示例,其中为简单起见,我们假设每个实心黑色边缘具有分数1,并且仅存在于第一或第二网络中的每个边缘分别具有分数0.5和0.2。考虑标记为a和b的节点。连接a和b的直接路径的得分为w(a,b)~1。节点a有3个通过守恒边连接的复合节点,有1个通过非守恒边连接的复合节点。节点B具有通过守恒边连接的3个复合节点,以及通过未配对的边连接的0个复合节点。因此,直接路径的贡献是:
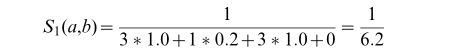
A和b评分之间有3条间接路径,分别为(0:2z0:2)~0:4、(0:5z0:5)~1、(0:2z1)~1:2,节点a有6条间接路径连接到其他组合节点,总分为7.6。节点B有7条间接路径将其连接到其他复合节点,总分为8.2。因此,i和j之间的间接路径的贡献为
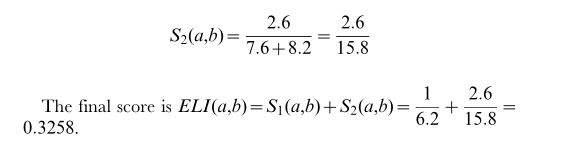
修剪并集图
由上述构造产生的对准图趋向于非常密集,其边分数分布在很大范围的值上。因此,在对准过程的下一步骤中,为了简化对齐图并降低计算成本,必须去除可靠性较低的边。查看边缘分数的分布时,会出现两个有趣的事实:
- 很少有边的得分明显高于其他边。
- 边分数在对齐图的不同区域之间差别很大,并且受拓扑特征(例如相互作用密度)的影响。因此,基于全局阈值修剪边可能不合适。
根据这两个观察结果,我们设计了一种剪枝策略,一次处理与同一节点关联的所有边,并且只保留局部高得分的边。使用了一个简单但有效的规则:
对于每个节点x[GA,设Eli(x,y?)~Maxy[N(X)(Eli(x,y))]。对于给定的常数t,所有边(x,y),y[N(X),得分为Eli(x,y)vtELi(x,y?)。都被删除了。
该修剪策略可通过改变阈值t来调整,从而允许创建更密集或更稀疏的网络。在我们的试验中,我们使用了t~0:5。修剪阈值t在0.3到0.7的范围内进行了测试,得到了相似的结果。这是意料之中的,因为入射到同一节点的高得分和低得分边之间的距离是尖锐的,如图5所示。另一方面,不修剪低得分边(t=0)会引入大量的虚假边。实际上,此过程的应用大大减少了对齐图的边数。
处理多个同源基因。同源关联通常是多对多的,与许多假定的同源基因相关的蛋白质将在比对图中显示为多个节点。当这些蛋白质被多次包含在同一溶液中时,这就变得至关重要,从而降低了最终图谱的准确性。
我们提出了一种策略,利用网络的拓扑结构来校正连接多个同源关联的节点的边的权重。假设y1,y2,??,yk,yi~(u,vi),是GA的节点,对应于同一节点u[V1,其中k个节点V1,?,V2的多个关联。此外,假设y1,y2,??,yk都与对齐图中的节点x相邻。我们希望在这些可能冲突的关联中识别最可能对应于与x的真实交互的关联。我们根据边(x,y1)、(x,y2)、::、(x,yk)的得分S(x,yi)对边进行排序,并用r(x,yi)表示边(x,yi)在排序列表中的等级。然后,我们通过将每个分数除以其排名来更正它:
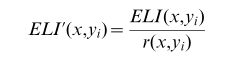
此修正降低了边的权重,得分最高的边不受影响。如上所述,我们在修剪边缘之前应用了此过程。我们观察到在解的质量和计算成本方面都有了显着的改进。为简单起见,在手稿的其余部分,我们将把这个修正后的分数称为Eli。
表4报告了针对S:Cerevisiae-D:Blackogaster和H:Sapiens-D:Blackogaster网络比对产生的比对图的统计数据。
表4.并集图和对齐图大小的统计数据
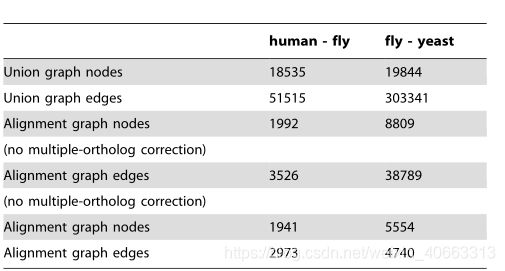
对于对齐图,考虑两种情况:当由于多个正交(如在我们的实验中)而存在对分配给边的权重的校正时,以及当不应用该校正时。
种子生成
种子由固定大小为k的比对图的一个小的子图组成,即k-子图。首先,从遗传算法中提取所有的k-子图,允许节点和边的任意重叠,然后选择不重叠的得分最高的子图作为种子,其余的只用于迭代扩展。我们在所有的实验中都设置了k~4。
枚举具有任意重叠的所有k个子图可能是耗时的,因为即使从稀疏网络中也可能提取大量的小的子图。为了优化提取过程,我们实现了一个简单的启发式算法,以避免对同一实例进行多次计数,从而使每个子图只被找到一次。确切地说,我们首先对图O:va?n的节点施加任意顺序,然后通过迭代地查看图中距离u小于k的节点NK(U)来提取包含节点u的所有子图,使得对于每个v[NK(U),O(V)Wo(U)。
我们基于每个k-子图组件(即节点和边)的单独分数来给每个k-子图分配分数。确切地说,给定对齐图GA的一个子图g,并分别用VA(G)和EA(G)表示该子图g的节点和边集,我们定义:

其中,w(K)表示两个相关蛋白质是同源的置信度,S(i,j)是如上定义的比对图中的边(i,j)的分数。
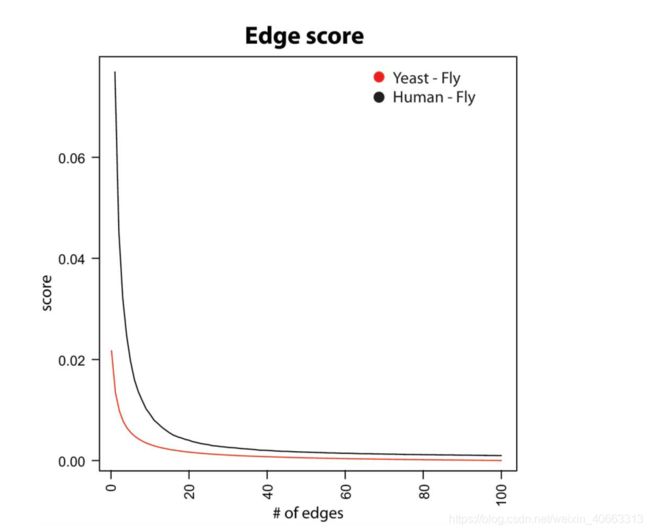
图5.关联到节点的边根据它们的得分进行排序。曲线上绘制的值是与节点关联的相同等级的边的分数在对齐图的所有节点上的平均值。为了具有可比较的值分布,我们选择至少有100条边的并集图上的所有节点。黑色曲线对应于具有1578个节点的人-蝇比对图,而红色曲线对应于具有9325个节点的酵母-苍蝇比对图。与对齐的网络无关,分数呈指数级递减,使得修剪步骤既重要又有效。
发现模块
一旦所有的k-子图都被提取和评分,算法就会根据它们的得分对它们进行排序,并选择得分最高的一个作为种子。该算法从种子开始,迭代地扩展候选解。该算法由若干扩展步骤组成。在每个扩展步骤中,与模块相邻的所有k-子图(即与其共享至少一个节点)被认为是扩展的候选。将满足特定要求的所有k-子图添加到模块,从而在每个步骤将一个或多个k-子图添加到当前模块。
选择要添加到模块中的k-子图是该方法的一个关键点,我们需要在这里提供一些定义。在下面,我们用IE(V)表示关联在节点v上的图GA的边集,用IEG(V)表示关联在节点v上的子图g的边集。最后,对于T的子集S,我们用T\S表示T的不在S中的元素的子集。给定当前模M、候选子图g和对齐图N~GA\Fm的剩余部分gg,关联在节点v[g]上的边集可以根据其被分成子集。正式地:
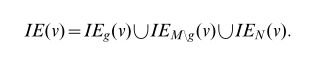
首先,我们定义了一个与模紧密相连的k-子图,如果![]()
紧连通子图始终添加到模块中。如果连接到模块的链路比连接到网络其余部分的链路更可靠,则连接松散连接的子图。
使用上面介绍的符号,对于给定的k-子图g,我们定义:
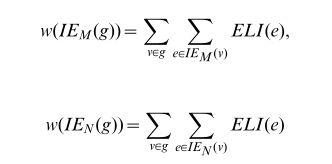
将g连接到模的边的权重之和,以及将g连接到网络的其余部分的边的权重的和。如果满足以下条件,则将g添加到模块中:
![]()
在扩展阶段结束时,所有接受的k-子图立即被添加到模块。重复这一过程,直到不能再添加更多的k-子图,因此我们不会对可获得的复合体的大小设置上限。另一方面,我们要求我们的解决方案至少有5个节点,种子的大小(4个节点)和至少要完成一个扩展步骤的要求施加了限制。值得注意的是,通过k-子图而不是一次一个节点来扩展模块不仅对方法的良好性能至关重要,而且也是考虑蛋白质与其直接邻居之间的多重依赖的关键。