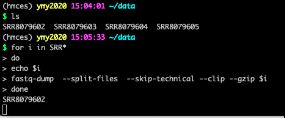
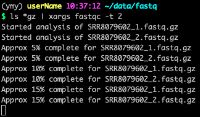
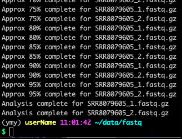
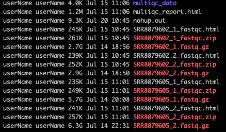
- 对人造子宫的忧虑——会不会出现异形?
怒火女子工坊
这个担忧,完全没有必要。人造子宫只是肉体子宫的替代品。打个比方,孵化器是给养殖场孵化鸡蛋的。和母鸡自己孵鸡蛋相比,它只是更方便而已,它并不能把鸡蛋变成鸭蛋、鹅蛋、鸵鸟蛋。同样的,人造子宫也不会改变人类幼体形状。是否异形,要看囊胚,那是基因生物学家的事儿,和人造子宫没有关系。科学家真能搞出异形来,在女性的肉体子宫里不能培育吗?人造子宫和异形不仅没有因果关系,也没有一点相关性。
- 中山司法亲子鉴定中心地址查询-13家(附2024年最新版)
国权基因
中山司法亲子鉴定中心地址在哪里?中山司法亲子鉴定咨询中心地址在中山市东区兴中道新兴花园翠园街(中山亲鉴生物)。中山亲子鉴定中心就是利用法医学、生物学和遗传学的理论和技术,从子代和亲代的形态构造或生理机能方面的相似特点,分析遗传特征,判断父母与子女之间是否是亲生关系,是法医物证鉴定的主要组成部分。很多人认为正规亲子鉴定需要在医院办理,这其实是一个误区,今天小编就来说说中山司法亲子鉴定中心地址在哪,供
- 重庆市12家正规合法个人隐私亲子鉴定机构位置一览(附2024最新鉴定中心汇总)
国医基因吴主任
在重庆市,亲子鉴定作为一项结合了医学、生物学与遗传学的科学技术,正逐渐成为解决家庭关系疑问的重要手段。亲子鉴定,又称亲权鉴定,其核心在于通过对人类遗传标记的深入检测与分析,科学而准确地判断父母与子女之间是否存在亲生关系。重庆正规司法鉴定机构汇总2.重庆法正司法鉴定所机构地址:重庆市渝中区公园路南区3.广东华曦法医物证司法鉴定所(重庆站)机构地址:重庆市沙坪坝区融汇新时代4.重庆市明正司法鉴定所机构
- 崔律·100天挑战阅读《奇特的一生》【《奇特的一生》·Day57·如何评价自己的一生(1)
冬夜读书YHY
这是“崔律精时力之100天挑战《奇特的一生》阅读”系列,今天是2019年3月30日一个人在离开了这个世界上以后。后人对他的评价是什么样子,在某种程度上也就决定了这个人在他的有生之年对这个世界做出的贡献吧。柳翁一生有一件非常固执的事情,就是长达几十年的时间记录时间统计时间计划。因为他的这种非常近乎于苛刻的这种方式,让他获得了很多的成就。同时,以它在生物学上的一些贡献,让他的后来的后继者会记住。会沿着
- 【农业模型】GPT地学领域应用、AquaCrop、R贝叶斯生态、Copula函数、DSSAT、APSIM、WOFOST、DNDC、CERRES、SWACRO、RZWQM、POTATO、SOLANUM
weixin_贾
遥感数据与作物模型地理遥感生态模型ChatGPT4/DeepSeekgptr语言无人机经验分享
农业模型依据研究对象分为农业生物模型、农业环境模型、农业技术模型、农业经济模型作物模型,即作物生长模拟模型(或称作物生长模型),是从系统科学的角度,基于作物生理过程机制,将气候、土壤、作物品种和管理措施等对作物生长的影响因素作为一个整体系统的数值模拟系统。能够以特定时间步长对作物在单点尺度上生长发育的生物学参数以及作物产量进行动态模拟,定量化研究环境因子以及田间管理措施对作物生长发育的影响。通用型
- Python 生物信息学秘籍第三版(四)
绝不原创的飞龙
默认分类默认分类
原文:annas-archive.org/md5/9694cf42f7d741c69225ff1cf52b0efe译者:飞龙协议:CCBY-NC-SA4.0第十一章:生物信息学中的机器学习机器学习在许多不同的领域中都有应用,计算生物学也不例外。机器学习在该领域有着无数的应用,最古老且最为人熟知的应用之一就是使用主成分分析(PCA)通过基因组学研究种群结构。随着该领域的蓬勃发展,还有许多其他潜在的应
- 《共情的力量》——共情是宽容的生物学基础
小村庄81
这世界上没有人是完全一模一样的,我们并不会有着完全同样的想法和感受。事实上,我们绝大多数人的反应是如此的迥异,是共情使我们能彼此相处。即使把语言拿走,我们还是能通过彼此的眼神、面部肌肉的移动,以及手的触摸进行相互沟通,让我们能看透彼此的内心和灵魂。通过共情,让我们能看到彼此的共同点——渴望连接的内心和渴求理解的灵魂。共情为我们带来宽容,它会创造出对生命多样性的积极欣赏和持久尊重。001共情对宽容的
- 2023-01-05
图灵基因
Nature|重新优化突变负荷指导免疫治疗决策原创三千图灵基因2023-01-0509:55发表于江苏收录于合集#前沿分子生物学机制撰文:三千IF=69.504推荐度:⭐⭐⭐⭐⭐亮点:通过与肿瘤/非肿瘤组织配对测序结果对比发现,因为不正确地将胚系突变指定为肿瘤突变,仅肿瘤组织测序分析大大高估了TMB,特别是非欧洲血统的患者。基于回归分析,提出了一种以遗传特异性的方式重新校准肿瘤检测组的TMB值的方
- 基因地理学家-1
中信书院
《基因传》是普利策奖得主悉达多·穆克吉继医学普及畅销书《众病之王:癌症传》后的又一医学科普专著,讲述了基因理论的起源、发展与未来,对科学史上最具挑战与危险的概念进行了诠释。《基因传》可以看作一部反映基因发展史的传记,全书按照时间顺序展开故事情节,没有医学或生物学背景的读者不妨将其内容分为两个部分看,前一部分是基因的发现史,后一部分是基因技术的应用史。
- matlab画信号图方法,献给初学者:手把手教你绘制信号通路图
信号通路是指能将细胞外的分子信号经细胞膜传入细胞内发挥效应的一系列酶促反应通路。细胞信号通路图是科研研究过程中最常见也是最常用到的,如何绘制适合我们自己科研课题的信号通路图呢?可以试试pathwaybuildertool软件。这款软件简单易学,即便是零基础的同学,也可以做出漂亮的信号通路。1.首先,打开PathwayBuilderTool2.0软件,软件自带分子生物学会用到的基本元素,如不同的细胞
- 冷冻电镜重构的GPU加速破局:从Relion到CryoSPARC的并行重构算法
九章云极AladdinEdu
人工智能pytorch架构gpu算力机器学习自然语言处理深度学习
点击“AladdinEdu,同学们用得起的【H卡】算力平台”,H卡级别算力,按量计费,灵活弹性,顶级配置,学生专属优惠。一、冷冻电镜重构的算力困局随着单粒子冷冻电镜(cryo-EM)分辨率突破原子级别(<3Å),重构算法计算复杂度呈指数级增长。传统CPU集群处理百万级粒子数据集需数周时间,成为结构生物学研究的关键瓶颈。本文重点分析Relion和CryoSPARC两大主流软件在GPU并行化方面的技术
- 富集分析——GO、KEGG
ersanshi055
生信小菜鸟富集分析GOkegg
一、富集分析的基础认知在生物信息学研究领域,基因功能解析及通路阐释是众多分析流程中的关键环节,富集分析(EnrichmentAnalysis)是将基因或蛋白列表按照功能进行分类的统计方法,目的是找出在特定基因集中显著富集的功能类别或通路。通过这种方法,研究人员可以理解一组基因(如差异表达基因)在哪些生物学过程、分子功能或通路中代表。1.富集分析分类基因本体论富集分析(GeneOntologyEnr
- python 科研作图_Origin科研绘图
weixin_39525933
python科研作图
前言入了生物学的坑,狗狗们需要时不时的画一些图,看着别人高大上的图片,大家有没有好奇这些图片是怎么做出来的呢?就本狗狗来看(狗狗可能来自农村-_-,),现在铺天盖地的paper里的图,有些,当然本身就是照片啦,比如跑胶啊WB啊,有些是用R、python、或者matlab做的,那么对于不懂编程的狗狗来说,就需要利用一些趁手作图软件,也可以做出毫不逊色于前者的美图,常见的这类软件有origin,gra
- 合成生物学奇点:AI驱动CRISPR超进化工厂2025投产纪实
前言前些天发现了一个巨牛的人工智能免费学习网站,通俗易懂,风趣幽默,忍不住分享一下给大家。点击跳转到网站《合成生物学奇点:AI驱动CRISPR超进化工厂2025投产纪实》副标题:全球首座AI-BioFab落地深圳,蛋白质设计周期从3年压缩至11天,生物制造成本暴跌90%一、生物制造范式的历史性颠覆▶︎传统生物工程的三大世纪困局graphTDA[缓慢的试错循环]-->B[单基因改造耗时≥6个月]C[
- 面向高校的人工智能通识教育课程实验设计方案
武汉唯众智创
人工智能人工智能通识教育课程实验人工智能通识教育人工智能通识课程人工智能通识
一、前言2018年,教育部发布《高等学校人工智能创新行动计划》,明确提出“重视人工智能与计算机、控制、数学、统计学、物理学、生物学、心理学、社会学、法学等学科专业教育的交叉融合,探索‘人工智能+X’的人才培养模式”。过去,人工智能教育多集中于研究生阶段,本科生接触机会相对有限。2019年,教育部批准35所高校增设“人工智能”本科专业,这标志着人工智能正式纳入本科教育体系。如今,人工智能课程大多是计
- 重生学AI第十五集:学习非线性激活函数
背景知识激活是什么意思?“激活”一词来源于生物学神经系统,在人的大脑中,存在着大量的神经元。每个神经元在接收到足够强的刺激时,会被激活,产生电信号并传递给其他神经元。这些电信号在神经网络中层层流动,最终形成了大脑对外界信息的反应。神经元就等同于人工神经网络中的基本计算单元,每一个网络层都包含着许多这样的神经元,激活函数就是为了能够判断输入是否达到“激活”标准,达到激活标准,则会影响后续计算,反之,
- 合成生物学奇点:AI驱动CRISPR超进化工厂2025投产纪实
HeartException
人工智能
前言前些天发现了一个巨牛的人工智能免费学习网站,通俗易懂,风趣幽默,忍不住分享一下给大家。点击跳转到网站《合成生物学奇点:AI驱动CRISPR超进化工厂2025投产纪实》副标题:全球首座AI-BioFab落地深圳,蛋白质设计周期从3年压缩至11天,生物制造成本暴跌90%一、生物制造范式的历史性颠覆▶︎传统生物工程的三大世纪困局graphTDA[缓慢的试错循环]-->B[单基因改造耗时≥6个月]C[
- 稳转细胞株构建全攻略:从理论到实践的生物制药核心技术解析
北京义翘神州
稳转细胞株构建服务
为何稳转细胞株成为生物制药领域的“兵家必争之地”?稳转细胞株被称为“药物研发的基石”,是单克隆抗体、双抗、疫苗、ADC等生物制品规模化生产的核心环节,还是药物生产成本、效率和质量的关键决定因素。一株高产稳定的细胞株有可能直接缩短药物上市周期3-5年。稳定细胞株因其稳定性高,适用于各种研究应用,包括重组蛋白、抗体生产、结构生物学和功能性研究等。那么,如何打造一株理想的稳转细胞株呢?义翘神州在重组表达
- 通过交互式网页探索传输现象-AI云计算数值分析和代码验证
亚图跨际
AI人工智能云计算
传输过程涉及质量、动量和能量等物理量在各种系统中的基本运动和转移,主要分为动量传输、热量传输和质量传输,在工程、环境科学、生物学和物流等领域至关重要。传输过程是指物理量(如质量、动量和能量)在物理、化学、生物或工程系统中的移动和传递。这些过程是各种科学和工程领域的基础,主要分为三类:☁️AI云计算数值分析和代码验证传输过程的类型动量传输这涉及动量在运动介质(例如流体)中的传递。它对流体流动、沉降、
- 【EI会议征稿】东北大学主办第三届机器视觉、图像处理与影像技术国际会议(MVIPIT 2025)
诗远Yolanda
图像处理计算机视觉考研视频机器学习论文阅读
一、会议信息大会官网:www.mvipit.org官方邮箱:
[email protected]会议地点:辽宁沈阳主办单位:东北大学会议时间:2025年9月27日-9月29日二、征稿主题集中但不限于“机器视觉、图像处理与影像技术”等其他相关主题。机器视觉:视觉中的统计机器学习;立体视觉标定;几何建模与处理;人脸识别与手势识别;早期视觉和生物学启发的视觉;光流法和运动追踪;图像分割和图像分类;基于模型的视觉
- CARLsim开源程序 是一个高效、易用、GPU 加速的软件框架,用于模拟具有高度生物细节的大规模脉冲神经网络 (SNN) 模型。
struggle2025
神经网络人工智能深度学习
一、软件介绍文末提供程序和源码下载CARLsim是一个高效、易用的GPU加速库,用于模拟具有高度生物学细节的大规模脉冲神经网络(SNN)模型。CARLsim允许在通用x86CPU和标准现成GPU上以逼真的突触动力学执行Izhikevich脉冲神经元网络。该模拟器在C/C++中提供了一个类似PyNN的编程接口,允许在突触、神经元和网络级别指定详细信息和参数。二、CARLsim6的新功能包括:CUDA
- AI助力基因遗传疾病检测:现状与未来
t0_54program
大数据与人工智能人工智能个人开发
在现代医学领域,与基因紊乱相关疾病的早期检测至关重要。像肺癌,早期诊断的患者5年生存率可达57%,而四期癌症患者生存率仅3%。阿尔茨海默病的早期检测,能让患者改变生活方式、参与临床试验并提前治疗脑部退化症状,有效延长生命。尽管基因检测对评估晚发性阿尔茨海默病的可能性有帮助,对早发性阿尔茨海默病也有指示作用,但其检测技术仍有待完善。目前,仅基于生物学研究的疾病检测技术多样,虽对特定病例精确,但通常需
- python做生物信息学分析_Python从零开始第五章生物信息学①提取差异基因
吴敬欣
python做生物信息学分析
目前来说,做生物信息学的人越来越多,但是我觉得目前而言做生信的主要有三类人:老本行是做实验的,做生信可能是为了辅助研究或者是为了发paper(有非常多的临床生选择趟生信这波水)主要是做生信的,主要涵盖高通量测序数据分析,组学数据分析等等,专门从事生物学数据分析的这群人,其大部分也是本科生物狗作为强大的生力军,以调包写R,python为主。那么这群人就要熟悉看各种包的tutorial以及如何进行常规
- 用Python实现生信分析——功能预测详解
写代码的M教授
生信分析python开发语言
功能预测是生物信息学中的一项重要任务,通过分析基因或蛋白质序列的特征,推测它们的生物学功能。功能预测通常涉及多种方法,包括序列比对、基序识别、机器学习模型等。这些方法可以帮助科学家推断未知基因的功能,从而加速生物学研究的进展。1.功能预测的主要方法(1)同源性比对:通过将未知基因或蛋白质序列与数据库中的已知序列进行比对,识别出同源序列,并推测它们的功能。常用工具包括BLAST、HMMER等。(2)
- 博弈论概述
C7211BA
博弈论
博弈论(GameTheory)是研究理性决策者在策略互动中如何行动和决策的数学理论。它广泛应用于经济学、政治学、生物学、计算机科学等领域。以下是博弈论的主要思想和核心概念:1.核心思想博弈论的核心是分析多个参与者(玩家)在相互依赖的情境中如何做出最优决策,即每个人的收益不仅取决于自己的选择,还取决于他人的选择。主要特点包括:策略互动:玩家的决策相互影响。理性假设:玩家追求自身利益最大化(理性人假设
- 13、动态边缘检测与人机交互:迈向更智能的未来
csp1223
机器人世界的探索与创新动态边缘检测人机交互视觉系统
动态边缘检测与人机交互:迈向更智能的未来1.引言随着机器人技术的快速发展,人机交互变得越来越重要。机器人不仅需要具备感知周围环境的能力,还需要能够理解和响应人类的行为。视觉系统是机器人与外界交互的关键组成部分,尤其是在边缘检测方面,它是衡量视觉系统性能的重要指标之一。传统的边缘检测方法大多基于静态掩码,这种方法虽然有效但在处理复杂环境时表现有限。近年来,研究人员借鉴生物学原理,尤其是视网膜的功能,
- 人工神经网络:单层神经网络(感知器)
一、神经网络介绍1、生物学起源与基本概念(1)生物神经网络启发人类大脑由约860亿个神经元组成,这些神经元通过突触相互连接,形成复杂网络。当外界刺激传入时,神经元会传递电信号并释放化学物质(神经递质),从而实现信息处理。人工神经网络正是模仿这一机制,通过数学模型构建“人工神经元”和“连接权重”。(2)人工神经网络的定义:由大量人工神经元(节点)相互连接构成的计算系统,通过调整节点间的连接权重来学习
- 10、生存时间分析:理论与实践
seiji morisako
生存时间分析删失数据生存函数
生存时间分析:理论与实践1.引言在现代数据分析中,生存时间分析(SurvivalAnalysis)是一个重要的分支,尤其在医学、生物学和工程学等领域中有着广泛应用。生存时间分析的核心问题是如何处理和分析那些随时间变化的事件,例如患者的存活时间、机器的使用寿命等。这类分析的一个关键挑战是如何处理研究过程中受试者的退出(censoring),即在研究结束前某些受试者可能已经不再参与研究,但他们并没有经
- 理解与建模弹性膜-AI云计算数值分析和代码验证
亚图跨际
AI人工智能云计算
弹性膜在连接生物学理解和工程创新方面至关重要,因为它们能够模拟软组织力学、实现先进的细胞培养系统和促进柔性设备,广泛应用于软组织生物力学、细胞培养、生物膜建模和生物医学工程等领域。☁️AI云计算数值分析和代码验证弹性膜在连接生物学理解和工程创新方面至关重要,其应用范围从模拟软组织力学到实现先进的细胞培养系统和柔性设备。它们的价值在于能够复制复杂的机械行为,并为生物医学和技术进步提供功能平台。以下是
- 【Python】BioPython
宅男很神经
python开发语言
第一章:万物之始——BioPython的世界观与基石欢迎来到分子生物学的数字新纪元。在这里,生命的蓝图——DNA、RNA和蛋白质——不再仅仅是湿实验(wetlab)中的化学实体,它们被转化为海量的、以GB乃至TB计的数字信息。要在这片数据的汪洋中航行,我们需要一艘坚固、强大且灵活的旗舰。在Python的世界里,这艘旗舰的名字,就是BioPython。本章,我们将为这趟漫长的远征奠定基石。我们不会满
- 微信开发者验证接口开发
362217990
微信 开发者 token 验证
微信开发者接口验证。
Token,自己随便定义,与微信填写一致就可以了。
根据微信接入指南描述 http://mp.weixin.qq.com/wiki/17/2d4265491f12608cd170a95559800f2d.html
第一步:填写服务器配置
第二步:验证服务器地址的有效性
第三步:依据接口文档实现业务逻辑
这里主要讲第二步验证服务器有效性。
建一个
- 一个小编程题-类似约瑟夫环问题
BrokenDreams
编程
今天群友出了一题:
一个数列,把第一个元素删除,然后把第二个元素放到数列的最后,依次操作下去,直到把数列中所有的数都删除,要求依次打印出这个过程中删除的数。
&
- linux复习笔记之bash shell (5) 关于减号-的作用
eksliang
linux关于减号“-”的含义linux关于减号“-”的用途linux关于“-”的含义linux关于减号的含义
转载请出自出处:
http://eksliang.iteye.com/blog/2105677
管道命令在bash的连续处理程序中是相当重要的,尤其在使用到前一个命令的studout(标准输出)作为这次的stdin(标准输入)时,就显得太重要了,某些命令需要用到文件名,例如上篇文档的的切割命令(split)、还有
- Unix(3)
18289753290
unix ksh
1)若该变量需要在其他子进程执行,则可用"$变量名称"或${变量}累加内容
什么是子进程?在我目前这个shell情况下,去打开一个新的shell,新的那个shell就是子进程。一般状态下,父进程的自定义变量是无法在子进程内使用的,但通过export将变量变成环境变量后就能够在子进程里面应用了。
2)条件判断: &&代表and ||代表or&nbs
- 关于ListView中性能优化中图片加载问题
酷的飞上天空
ListView
ListView的性能优化网上很多信息,但是涉及到异步加载图片问题就会出现问题。
具体参看上篇文章http://314858770.iteye.com/admin/blogs/1217594
如果每次都重新inflate一个新的View出来肯定会造成性能损失严重,可能会出现listview滚动是很卡的情况,还会出现内存溢出。
现在想出一个方法就是每次都添加一个标识,然后设置图
- 德国总理默多克:给国人的一堂“震撼教育”课
永夜-极光
教育
http://bbs.voc.com.cn/topic-2443617-1-1.html德国总理默多克:给国人的一堂“震撼教育”课
安吉拉—默克尔,一位经历过社会主义的东德人,她利用自己的博客,发表一番来华前的谈话,该说的话,都在上面说了,全世界想看想传播——去看看默克尔总理的博客吧!
德国总理默克尔以她的低调、朴素、谦和、平易近人等品格给国人留下了深刻印象。她以实际行动为中国人上了一堂
- 关于Java继承的一个小问题。。。
随便小屋
java
今天看Java 编程思想的时候遇见一个问题,运行的结果和自己想想的完全不一样。先把代码贴出来!
//CanFight接口
interface Canfight {
void fight();
}
//ActionCharacter类
class ActionCharacter {
public void fight() {
System.out.pr
- 23种基本的设计模式
aijuans
设计模式
Abstract Factory:提供一个创建一系列相关或相互依赖对象的接口,而无需指定它们具体的类。 Adapter:将一个类的接口转换成客户希望的另外一个接口。A d a p t e r模式使得原本由于接口不兼容而不能一起工作的那些类可以一起工作。 Bridge:将抽象部分与它的实现部分分离,使它们都可以独立地变化。 Builder:将一个复杂对象的构建与它的表示分离,使得同
- 《周鸿祎自述:我的互联网方法论》读书笔记
aoyouzi
读书笔记
从用户的角度来看,能解决问题的产品才是好产品,能方便/快速地解决问题的产品,就是一流产品.
商业模式不是赚钱模式
一款产品免费获得海量用户后,它的边际成本趋于0,然后再通过广告或者增值服务的方式赚钱,实际上就是创造了新的价值链.
商业模式的基础是用户,木有用户,任何商业模式都是浮云.商业模式的核心是产品,本质是通过产品为用户创造价值.
商业模式还包括寻找需求
- JavaScript动态改变样式访问技术
百合不是茶
JavaScriptstyle属性ClassName属性
一:style属性
格式:
HTML元素.style.样式属性="值";
创建菜单:在html标签中创建 或者 在head标签中用数组创建
<html>
<head>
<title>style改变样式</title>
</head>
&l
- jQuery的deferred对象详解
bijian1013
jquerydeferred对象
jQuery的开发速度很快,几乎每半年一个大版本,每两个月一个小版本。
每个版本都会引入一些新功能,从jQuery 1.5.0版本开始引入的一个新功能----deferred对象。
&nb
- 淘宝开放平台TOP
Bill_chen
C++c物流C#
淘宝网开放平台首页:http://open.taobao.com/
淘宝开放平台是淘宝TOP团队的产品,TOP即TaoBao Open Platform,
是淘宝合作伙伴开发、发布、交易其服务的平台。
支撑TOP的三条主线为:
1.开放数据和业务流程
* 以API数据形式开放商品、交易、物流等业务;
&
- 【大型网站架构一】大型网站架构概述
bit1129
网站架构
大型互联网特点
面对海量用户、海量数据
大型互联网架构的关键指标
高并发
高性能
高可用
高可扩展性
线性伸缩性
安全性
大型互联网技术要点
前端优化
CDN缓存
反向代理
KV缓存
消息系统
分布式存储
NoSQL数据库
搜索
监控
安全
想到的问题:
1.对于订单系统这种事务型系统,如
- eclipse插件hibernate tools安装
白糖_
Hibernate
eclipse helios(3.6)版
1.启动eclipse 2.选择 Help > Install New Software...> 3.添加如下地址:
http://download.jboss.org/jbosstools/updates/stable/helios/ 4.选择性安装:hibernate tools在All Jboss tool
- Jquery easyui Form表单提交注意事项
bozch
jquery easyui
jquery easyui对表单的提交进行了封装,提交的方式采用的是ajax的方式,在开发的时候应该注意的事项如下:
1、在定义form标签的时候,要将method属性设置成post或者get,特别是进行大字段的文本信息提交的时候,要将method设置成post方式提交,否则页面会抛出跨域访问等异常。所以这个要
- Trie tree(字典树)的Java实现及其应用-统计以某字符串为前缀的单词的数量
bylijinnan
java实现
import java.util.LinkedList;
public class CaseInsensitiveTrie {
/**
字典树的Java实现。实现了插入、查询以及深度优先遍历。
Trie tree's java implementation.(Insert,Search,DFS)
Problem Description
Igna
- html css 鼠标形状样式汇总
chenbowen00
htmlcss
css鼠标手型cursor中hand与pointer
Example:CSS鼠标手型效果 <a href="#" style="cursor:hand">CSS鼠标手型效果</a><br/>
Example:CSS鼠标手型效果 <a href="#" style=&qu
- [IT与投资]IT投资的几个原则
comsci
it
无论是想在电商,软件,硬件还是互联网领域投资,都需要大量资金,虽然各个国家政府在媒体上都给予大家承诺,既要让市场的流动性宽松,又要保持经济的高速增长....但是,事实上,整个市场和社会对于真正的资金投入是非常渴望的,也就是说,表面上看起来,市场很活跃,但是投入的资金并不是很充足的......
- oracle with语句详解
daizj
oraclewithwith as
oracle with语句详解 转
在oracle中,select 查询语句,可以使用with,就是一个子查询,oracle 会把子查询的结果放到临时表中,可以反复使用
例子:注意,这是sql语句,不是pl/sql语句, 可以直接放到jdbc执行的
----------------------------------------------------------------
- hbase的简单操作
deng520159
数据库hbase
近期公司用hbase来存储日志,然后再来分析 ,把hbase开发经常要用的命令找了出来.
用ssh登陆安装hbase那台linux后
用hbase shell进行hbase命令控制台!
表的管理
1)查看有哪些表
hbase(main)> list
2)创建表
# 语法:create <table>, {NAME => <family&g
- C语言scanf继续学习、算术运算符学习和逻辑运算符
dcj3sjt126com
c
/*
2013年3月11日20:37:32
地点:北京潘家园
功能:完成用户格式化输入多个值
目的:学习scanf函数的使用
*/
# include <stdio.h>
int main(void)
{
int i, j, k;
printf("please input three number:\n"); //提示用
- 2015越来越好
dcj3sjt126com
歌曲
越来越好
房子大了电话小了 感觉越来越好
假期多了收入高了 工作越来越好
商品精了价格活了 心情越来越好
天更蓝了水更清了 环境越来越好
活得有奔头人会步步高
想做到你要努力去做到
幸福的笑容天天挂眉梢 越来越好
婆媳和了家庭暖了 生活越来越好
孩子高了懂事多了 学习越来越好
朋友多了心相通了 大家越来越好
道路宽了心气顺了 日子越来越好
活的有精神人就不显
- java.sql.SQLException: Value '0000-00-00' can not be represented as java.sql.Tim
feiteyizu
mysql
数据表中有记录的time字段(属性为timestamp)其值为:“0000-00-00 00:00:00”
程序使用select 语句从中取数据时出现以下异常:
java.sql.SQLException:Value '0000-00-00' can not be represented as java.sql.Date
java.sql.SQLException: Valu
- Ehcache(07)——Ehcache对并发的支持
234390216
并发ehcache锁ReadLockWriteLock
Ehcache对并发的支持
在高并发的情况下,使用Ehcache缓存时,由于并发的读与写,我们读的数据有可能是错误的,我们写的数据也有可能意外的被覆盖。所幸的是Ehcache为我们提供了针对于缓存元素Key的Read(读)、Write(写)锁。当一个线程获取了某一Key的Read锁之后,其它线程获取针对于同
- mysql中blob,text字段的合成索引
jackyrong
mysql
在mysql中,原来有一个叫合成索引的,可以提高blob,text字段的效率性能,
但只能用在精确查询,核心是增加一个列,然后可以用md5进行散列,用散列值查找
则速度快
比如:
create table abc(id varchar(10),context blog,hash_value varchar(40));
insert into abc(1,rep
- 逻辑运算与移位运算
latty
位运算逻辑运算
源码:正数的补码与原码相同例+7 源码:00000111 补码 :00000111 (用8位二进制表示一个数)
负数的补码:
符号位为1,其余位为该数绝对值的原码按位取反;然后整个数加1。 -7 源码: 10000111 ,其绝对值为00000111 取反加一:11111001 为-7补码
已知一个数的补码,求原码的操作分两种情况:
- 利用XSD 验证XML文件
newerdragon
javaxmlxsd
XSD文件 (XML Schema 语言也称作 XML Schema 定义(XML Schema Definition,XSD)。 具体使用方法和定义请参看:
http://www.w3school.com.cn/schema/index.asp
java自jdk1.5以上新增了SchemaFactory类 可以实现对XSD验证的支持,使用起来也很方便。
以下代码可用在J
- 搭建 CentOS 6 服务器(12) - Samba
rensanning
centos
(1)安装
# yum -y install samba
Installed:
samba.i686 0:3.6.9-169.el6_5
# pdbedit -a rensn
new password:123456
retype new password:123456
……
(2)Home文件夹
# mkdir /etc
- Learn Nodejs 01
toknowme
nodejs
(1)下载nodejs
https://nodejs.org/download/ 选择相应的版本进行下载 (2)安装nodejs 安装的方式比较多,请baidu下
我这边下载的是“node-v0.12.7-linux-x64.tar.gz”这个版本 (1)上传服务器 (2)解压 tar -zxvf node-v0.12.
- jquery控制自动刷新的代码举例
xp9802
jquery
1、html内容部分 复制代码代码示例: <div id='log_reload'>
<select name="id_s" size="1">
<option value='2'>-2s-</option>
<option value='3'>-3s-</option