【PANDA教程】PANDA详细教程笔记
前 言
PANDA是一个基于FSL的DTI数据分析软件,从安装Linux系统开始到学习DTI的分析方法几乎花了一个月时间,但其实一半的时间都是在解决系统和软件的问题,当平台搭建好了以后,个人感觉DTI数据的分析相对于fMRI来说更简单易懂一些,因为构建结构网络的步骤相对更少,同时PANDA的出错率也相对较低,但输出的文件特别多,因此整理了一下笔记以便今后查看。该笔记主要是参照官方的视频教程和说明书整理的,里面还会补充一些自己在操作过程中的想法、笔记和注意事项等等。
Linux系统:Ubuntu 18.04.1
MATLAB_Linux版本:2018a
FSL官网:https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/
PANDA官方软件及教程:https://www.nitrc.org/frs/?group_id=582
FSL需要在Linux环境下使用,因此使用PANDA前需要在Linux系统中事先安装好FSL和Linux版本的MATLAB,这两个软件的安装教程在博客上有很多优秀的分享可供参考。
目录
一、数据准备
二、基本操作流程
Part 1 路径选择和基本设置(1~4)
1. DICOM/NIFTI Path文件输入
2. Result Path 设置结果输出路径
3. Subject_IDs 设置样本编号
4. File_Prefix 设置文件名前缀
Part 2 参数设置(5~7)
5. PiPeline Opt 设置并行处理参数
6. Diffusion_Opt 弥散参数设置(重点)
7. Tracking_Opt 纤维追踪及网络参数设置(重点)
Part 3 其他设置(8~13)
8. Load
9. RUN
10. Failed jobs
11. Clear
12. Terminate jobs
13. QUIT
三、输出结果
Part 1 样本基本信息
Part 2 结果文件(重点)
1. 基于图谱水平分析的结果
2. 基于TBSS的分析以及TBSS与图谱结合的分析结果
3. 基于体素的分析结果
Part 3 质量控制文件 quality_control
安装好所有软件后,在MATLAB中输入大写“PANDA”,回车,即能弹出以下操作界面:

一、数据准备
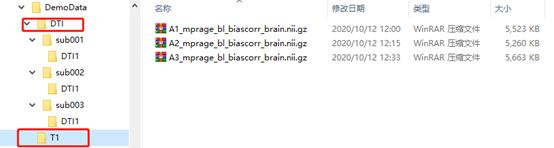
图像可以是DICOM或NIFTI格式,文件结构如下图所示,PANDA这个软件只需要文件结构统一即可,文件的名称没有特别限制。需要注意两点:
(1)DTI数据:每个sub如果仅扫描了一次,则sub文件夹下只需要包含一个DTI1文件夹(注意仅扫描了一次也一定要有这个文件夹,不然会出现The input for the 1subject is illegal),如果扫描了两次,则需要同时包含DTI1和DTI2两个文件夹,扫描多次的样本以此类推。另外,如果DTI数据是NIFTI格式,每个文件夹需要同时包含三个文件:“.bval”文件、“.bvec文件”和“.nii.gz文件”。其中“.bval”文件表示b value,里面存放的是每个扫描方向的b值,一般第一个b=0,后面的b常见的有1000,2400等;“.bvec文件”表示b vector,里面存放是每个扫描方向的x,y,z方向向量,三个方向的平方和应该为1。
(2)T1数据:T1数据仅需要把所有样本按照顺序放在同一个文件夹下即可。要注意T1图像的样本顺序要和前面DTI图像的样本顺序一致。

二、基本操作流程
Part 1 路径选择和基本设置(1~4)

1. DICOM/NIFTI Path文件输入
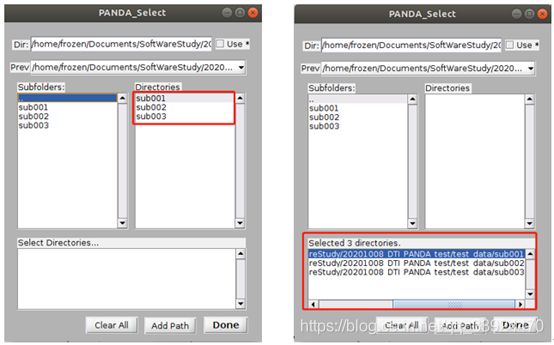
选中DTI的文件路径后,在右边的文件夹按下Shift的同时点击最后一个文件夹sub003,即可同时选择所有样本数据。在选择好所有数据后,在PANDA的蓝色框框处会自动显示所有样本的路径。
2. Result Path 设置结果输出路径

自己首先新建一个RESULT文件夹,然后选中该文件夹作为结果输出路径即可。
3. Subject_IDs 设置样本编号
在空格处按照matlab的命令格式输入ID号,如这里有三个样本,可直接输入[1:3](MATLAB的命令格式),然后回车即可。
4. File_Prefix 设置文件名前缀
如果需要,可以给结果文件加一个统一的文件名前缀,在此空格处输入即可。
Part 2 参数设置(5~7)

5. PiPeline Opt 设置并行处理参数

① 使用单台机器,点击batch,后面的Max_Queued的数字代表机器的CPU数,自己的机器有多少个CPU就填多少,但是DTI数据处理起来的数据运算量比较大,如果在跑数据之余还需要用电脑处理做其他的任务,建议留出来一个CUP,免得机器卡死,即如果自己电脑有8个核,这里可以填7个核即可,剩下一个核用于处理其他的任务,当然也可以按照自己需要进行填写。

② 如果在SGE的集群环境下,可以点击qsub,后面的Max_Queued同样表示集群环境下的总核数。
6. Diffusion_Opt 弥散参数设置 (重点)
弥撒参数设置分为两部分,第一部分为预处理参数设置,第二部分为弥散矩阵的参数设置。
(1)Preprocessing 预处理参数设置

- ① Resampling_Resolution
设置重采样的分辨率,默认是不用做的,因为一般机器采集出来的数据分辨率都是[2 2 2]mm的,但有的机器采集出来的数据分辨率是[1 1 1 ]或[0.9 0.9 1]等等,这些较高分辨率对原始结构图象的意义不是特别大,同时会增加很多计算量(尤其在计算纤维追踪时),此时较高分辨率的图像一般需要重采样到[2 2 2]。
- ② Orientation Path
数据采集时候会涉及到多个方向和多个角度,机器在采集数据时会有一个专门的坐标系,而FSL在分析数据时也会有一个坐标系,当数据采集时的坐标系和FSL设定的坐标系不一致时,就需要利用该参数进行调整。现在一般数据的采集坐标系和FSL的坐标系都是一致的,因此一般不用进行调整。如果想要知道数据采集的坐标系和FSL的坐标系是否一致,可以使用PANDA自带的Test Bvecs工具进行测试,若不一致就需要进行调整。一般可能出现的问题是需要调整invert,调整三个方向中的其中一个方向。
- ③ Raw NII from Dicon
选择keep/delete表示保留/删除由原始DICOM数据转换出来的NIFTI数据。如果原始数据为DICOM时,为了增加存储空间可以选择删除转换出来的NIFTI数据;如果原始数据本身就是NIFTI时,选择keep/delete都不会删除原始的NIFTI数据。
- ④ f(skull_removal) & Cropping gap
f(skull_removal):对DTI数据进行去头皮操作的参数,该参数越大表示去除的头皮越多,留下的大脑部分越少,一般默认使用0.25即可。
Cropping gap:在去除头皮后,图像会有很大一部分是没有信息的,如下图所示,大脑外面黑色的一大圈都是没用的,如果把整个图像进行计算的话会增加许多额外的计算量,因此这一步的功能其实就是把外面没用的那一圈截掉,以增加数据处理速度。而该参数的数值则表示大脑到截取框的距离,一般默认为3mm,如下图所示:
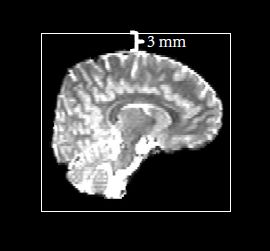 图片出自官方说明书
图片出自官方说明书
(2)Producing diffusion metrics 弥散矩阵参数设置

- ① Local Diffusion Homogeneity (LDH)
LDH计算的是某一个体素弥散率序列和邻接体素弥散率序列之间的相关性。PANDA分别使用kendall系数和spearman系数来计算这种相关性。该参数是是用于定义邻近点的,相邻的定义方式有三种,分别为面相邻,边相邻和点相邻,其中7 voxels表示面相邻(6个邻接体素+自己),19 voxels表示边相邻(18个邻接体素+自己),27 voxels表示点相邻(26个邻接体素+自己)。
- ② Normalizing_Targer
选择标准化模板。在计算出FA,MD,AD,RD等diffusion参数以后,需要把它们配准到标准模板,这里默认选择的是FSL提供的一个分辨率为1x1x1mm的标准FMRIB58_FA模板,可以根据自己需要下载或选择其他的标准模板,该标准模板如下图所示。

- ③ Smooth
对全脑进行voxel级别的分析前,需要先对diffusion参数进行重采样和平滑,以减少配准误差。
Normalizing_Resolution:注意,这里的重采样指的是对FA,MD,AD,RD等diffusion参数进行重采样(因为前面的标准FA模板的分辨率是1x1x1的,因此这里需要把这些diffusion参数重采样成2x2x2),而前面Preprocessing中的重采样指的是对整幅原始图像进行重采样。
Smoothing_kernel:平滑参数,FWHM,一般使用默认参数6即可。
- ④ Applying TBSS
在默认情况下是没有勾选TBSS(基于纤维束追踪的空间统计分析)的,也就是一般情况下不会在这个界面选择做TBSS,因为TBSS的计算过程中需要计算所有样本的平均骨架,如果在被试数据还没有采集完的情况下,做TBSS是没有意义的,除非已经确定需要的样本数据都已经全部采集完,才可以直接在这里选择做TBSS。因此在数据未采集完全时,可以先不在这里选择TBSS,等数据全部采集完全后,PANDA的Utilities中专门把TBSS工具箱单独分了出来,可以采用单独的TBSS工具箱进行分析。
Skeleton_cutoff:TBSS在纤维追踪时的停止参数,一般默认FA<0.2时停止追踪。
- ⑤ Atlas
选择白质分区图谱。标准分区图谱的存放位置为xxxx/PANDA_1.3.1_64/data/atlas/……。
WM_Label_Atlas:这里选择的图谱是通过手工分割得到的分区图谱,默认使用的是rICBM_DTI_81_WMPM_FMRIB58.nii.gz标准模板,共有50个分区,该图谱如下图所示:
 图片出自官方说明书
图片出自官方说明书
WM_Probtract_Atlas:Protract表示Prob-tract,即Probability-tract,这里选择的图谱是通过确定性纤维追踪方法跑出来的,并对所有样本结果进行平均概率识别的概率图谱。这里默认使用JHU_ICBM_tracts_maxprob_thr25_1mm.nii.gz标准模板,共有20个分区,该图谱如下图所示:
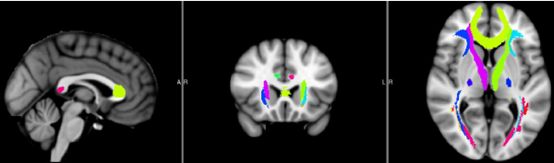 图片出自官方说明书
图片出自官方说明书
标准模板可根据自己研究需要进行选择。
若仅需要计算每个样本的FA,MD,AD,RD指标,而不需要构建结构网络时,可跳过下面的网络参数设置,直接查看 “Part 3 其他设置 ”,或直接点击RUN执行,开始计算 Diffusion 指标。
7. Tracking_Opt 纤维追踪及网络参数设置(重点)
纤维追踪参数设置分为三部分,分别为确定性纤维追踪参数设置、网络节点定义以及网络构建。这三部分主要用于追踪白质纤维和计算结构网络,默认情况下这一部分是全空的,可以根据个人研究课题选择自己需要分析的部分进行参数设置即可。
(1)Deterministic Fiber Tracking 纤维追踪参数设置

这一部分的作用是追踪出全脑的白质纤维。
- ① Propagation_Algorithm
选择纤维追踪算法,一般默认选择FACT(Fiber Assignment by Continuous Tracking)方法,后面还可以选择其他的纤维追踪算法,如下图所示:

这几种纤维追踪方法是参考Diffusion Toolkit软件来做的,除查找相关文献外,还可以到Diffusion Toolkit的官网上去了解。
Diffusion Toolkit官网:http://trackvis.org/dtk/
- ② Angle_Threshold & FA_Treshold
Angle_Threshold:在纤维追踪的时,在两次追踪之间的转角大于该阈值时就停止追踪,默认为45°
FA_Treshold:在纤维追踪时,当FA不在该阈值范围内时停止追踪,默认FA取0.2~1。
以上两个参数的示意图如下:
 图片出自官方教程
图片出自官方教程
- ③ Step_Length
纤维追踪过程中两步之间的步长。如果在中选择的纤维追踪算法是FACT时,不需要用到该参数,而当选择另外三种纤维追踪算法时才需要设置,PANDA已经设置好每个算法默认的Step_Length,直接使用默认值即可
- ④ Random_Seed
设置每个体素的种子点。在纤维追踪时都是先从一个种子点出发一步一步进行追踪,在一个体素内可以同时设置多个种子点,一般情况下都是一个体素设置一个种子点,也就是该体素的中心位置(默认设置)。若一个体素中设置了n个种子点,那么会在该体素中随机生成n个位置作为种子点,这里根据自己需要进行设置即可。
- ⑤ Apply Sline Filter
纤维追踪时都是从一个体素直线到另一个体素,因此这一步是对纤维追踪结果进行平滑。
(2)Network Node Definition 网络节点定义

网络节点和边的定义:使用灰质脑图谱对大脑进行分割后,每一个脑区作为一个网络节点,而上面部分追踪出来的白质纤维作为网络的边。因此在构建结构网络时同时需要每个样本的T1结构图像(用于定义网络节点)和FA图像(用于定义边)。
- ① Parcellated (native space)
网络节点的定义需要用到每个个体T1图像的大脑分区图像。该选项是当自己已经有每个个体T1图像的大脑分区文件时,直接选进来即可。如果自己手里的T1数据是原始数据还未进行分割,那么就要进行下面②~④的步骤。
另外,如果第一次已经通过②~④定义过节点,在跑第二次时不需要再重新定义节点,只需要把上一次的节点定义结果文件从这里选进来即可。
- ② T1 Images & Atlas
T1 Images:选择每个样本的T1图像,把最开始数据准备时已经整理好的T1图像全部选进来即可,选择方法同样是按住Shift的同时,点击最后一个样本即能完成全选。

Atlas:选择T1图像的分区图谱,默认使用ALL_90图谱,可根据自己需要进行选择。
- ③ T1 Template
把T1图配准到标准MNI空间。
- ④ T1 Options
对T1图像进行预处理的参数设置。
f(skull_removal):去头皮,默认参数为0.5;
Cropping_Gap:截去T1图像外面没用的部分以节省空间和加快处理速度。这里的参数设置和前面 “6. 弥散参数设置(1)④” 是一样的,都表示图像到截图框的距离;
Resample_Resolution:把T1图像重采样到1x1x1;
注意:由于这里的预处理并没有去除脖子这一步,因此输入的T1图像必须是已经除去脖子的:
a/ 如下图这种没有去除脖子的T1图像会严重影响后面的配准质量,因此不建议直接输入这种T1图像,最好事先对该图像进行去除脖子的预处理。如果原始数据是DICOM格式,可以使用MRIcron软件把图像转成NIFTI,其中“co-”开头的NIFTI图像即为已经去除脖子的图像。

b/ 最好输入如下图这种已经去除脖子的T1图像:

c/ 当然,还可以直接输入已经提前做好了所有预处理(包括去头皮、去脖子、平滑等等)的T1图像,但要注意此时第④步的T1预处理参数也要进行相应修改,如不用再选f(full_removal)进行去头皮的操作。

- ⑤ Path of FA
输入FA图像用于定义边,FA图像的路径会自动生成,不需要自己选择。
(3)Network Construction 网络构建
用于构建网络,包括构建确定性网络和概率网络。

- ① Deterministic
直接构建确定性网络。
- ② Bedpost+Probabilistic
基于概率性纤维追踪构建的概率网络。点击后会出现上面的窗口,由于概率性纤维追踪首先会用Bedpostx在每个体素内生成一个概率分布函数,左边的Fibers/Weight/Burnin是该函数的相关参数,使用FSL推荐的默认参数即可。
Tracking Type中的OPD是指直接输出纤维路径的概率分布;PD会先按照纤维的长度对路径分布先做一个校正,再输出纤维路径的概率分布。一般常用OPD方法。
Label ID是这个窗口中唯一需要我们手动输入的参数。这里可以根据自己的研究需要输入相对应的脑区编号。如在AAL_90图谱中,每一个脑区都会有一个对应的编号,分别为1到90,当我们想研究第3,4,5,8脑区之间的网络连接时,那么我们只需要在框框内输入[3:5 8]即可(MATLAB命令格式)。
根据自己需要设置好以上所有参数以后,即可点击RUN执行程序,开始构建结构网络。
Part 3 其他设置(8~13)

8. Load
在每次进行参数设置以后,都会在结果文件夹中生成一个后缀名为“.PANDA”的配置文件,该文件非常非常重要,因为它不仅仅记录了所有的参数设置,还记录了PANDA已经运行到哪一个步骤。
注意:PANDA可以后台运行,即设置好参数点击RUN后,可以直接关掉PANDA界面或者MATLAB,这并不会影响程序的进行。因此如果想要查看程序已经运行到哪个步骤,可以重新打开PANDA,把“.PANDA”文件Load进来,即可在下面的黄色框框内看到每个样本的处理进度。
9. RUN
点击RUN后开始在后台执行程序。
10. Failed jobs
如果在黄色框框的Status中发现有“failed”报告时,可以点击该按钮打开一个FailedLogs,以查看在哪一步出现错误,还可以点击FailedLogs中的每一条信息来查看任务出错的具体原因,如下图所示:
 图片来自官方说明书
图片来自官方说明书
11. Clear
一键清除PANDA的所有设置。(如果已经开始执行程序,点击Clear也不会影响后台的任务执行。)
12. Terminate jobs
如果需要中止后台正在运行的程序,需要先把该任务的“.PANDA”文件Load进来,然后点击该按钮即可全部中止。
13. QUIT
退出PANDA,如果已经开始执行程序,点击QUIT也不会影响后台的任务执行。
三、输出结果
Part 1 样本基本信息

1. “.PANDA”配置文件;
2. 所有样本的扫描参数,包括Voxels,volumes等等;
3. 所有样本的输入-输出路径对应信息,如下图所示:
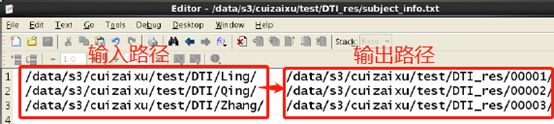 图片来自官方说明书
图片来自官方说明书
Part 2 结果文件(重点)
1. 基于图谱水平分析的结果
AllAtlasResults 文件夹

WMlabelResults是指基于手工分割白质图谱中50个分区的Diffution指标计算结果;WMtractResults使指白质概率图谱中20个分区的Diffution指标计算结果。
一共有6个Diffution指标:
- 06LDHs:LDH的计算结果(详细看第二节Part2的6(2)①),其中06表示邻近点的定义选择了面相邻,s表示该结果是通过spearman系数来计算相关的。(在计算spearman相关时不需要包括该像素点本身,所以是6。)
- 07LDHk:LDH的计算结果(详细看第二节Part2的6(2)①),其中07表示邻近点的定义选择了面相邻,k表示该结果是通过kendall系数来计算相关的。(在计算kendall相关时需要包括该像素点本身,所以是7。)
- FA:各向异性指数;
- L1:表示λ1,轴向弥散指数,即AD;
- L23:表示λ2和λ3的平均值,径向弥散指数,即RD;
- MD:平均弥散率。
注意以上6个Diffusion指标的标记形式,下面所有文件中的结果都用它们来指示不同的指标。
2. 基于TBSS的分析以及TBSS与图谱结合的分析结果

(1)仅基于TBSS的分析结果
Merged4DSkeleton文件夹:把所有样本的FA(或AD、RD、MD)骨架组合成一个4D的图像,可以直接用于统计分析,如下图所示:
 图片出自官方教程
图片出自官方教程
MeanData文件夹:TBSS生成的平均FA图像(FA)和平均FA骨架图像(FA_skeleton)以及它们对应的mask,其中最后一个“mask_dst”是计算骨架过程中产生的一个中间结果,不用管;
(2)TBSS与图谱结合的分析结果:
AllAtlasResults文件夹:TBSS方法追踪出白质纤维后,结合图谱的所有分析结果,具体每个文件的意思参照上一小节的说明;
3. 基于体素的分析结果
在结果文件夹中每个样本都会有一个单独的文件夹存放该样本所有的结果,每个样本文件夹打开后都是一样的。下面分别对一些重要的、我们需要用到的文件夹作一些说明。
(1) Standard_space文件夹

native_space是在样本原本空间中算出来的AF值等指标,standard_space则是把这些指标配准到标准空间后的结果,因此直接看standard_space中的结果即可。
打开standard_space有很多文件,如下图所示(仅展示部分):

- ① 黄色:表示每个Diffusion指标;
- ② 蓝色:1mm表示每个指标配准到分辨率为1x1x1标准空间后的结果;2mm表示分辨率重采样到2x2x2的结果;
- ③ 绿色:分别表示每个指标的图像和其骨架图像(skeletonised)在label图谱(WMlabel)和概率图谱(WMtract)分区中的计算结果。其中所有样本的骨架图像已经在前面TBSS - Merged4DSkeleton文件夹中整合成一个4D的nii文件,因此这里其实可以不用太关注;
- ④ 红色:s6mm表示平滑参数为[6 6 6];
分析时需要用到的图像为画红线的文件,也就是配准到标准空间后把分辨率重采样到2x2x2,并进行了平滑的图像。(具体参数设置详看第二节Part2——6.(2) ②~③)
(2)trackvis文件夹

该文件夹中存放的是纤维追踪的结果。其中第一个文件是纤维追踪的原始结果,第二个最后有“xxx_S.trk”的文件是经过平滑后的结果。
注意:前面两个“.trk”文件需要用TrackVis软件进行查看,该软件在Linux中只能通过命令行打开,结果如下图所示。
TrackVis下载网站:http://trackvis.org/download/
(3)Network文件夹

该文件夹中存放的是脑网络的构建结果,分别为确定性网络和概率网络。
Deterministic文件夹——
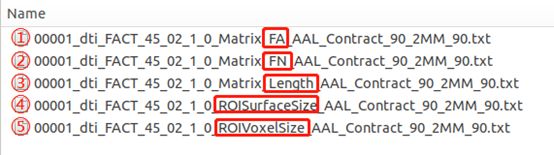
确定性网络的结果分别有(都是对称矩阵,如这里使用的是ALL_90图谱,因此这里的结果都是90x90的一个对称矩阵):
- FA:表示利用脑图谱每个分区之间的平均FA值构建的脑网络;
- FN:表示利用脑图谱每个分区之间的纤维数量构建的脑网络;
- Length:表示利用脑图谱每个分区之间的平均纤维长度构建的脑网络;
- ROISurfaceSize:表示每个脑区中,有纤维经过的体素有多少个;
- ROIVoxelSize:表示每个脑区有多少个体素;
确定性网络指的是前面三个网络,最后两个文件的信息是根据自己的研究需要,用于对前面三个网络进行校正处理的。
Probabilistic文件夹——
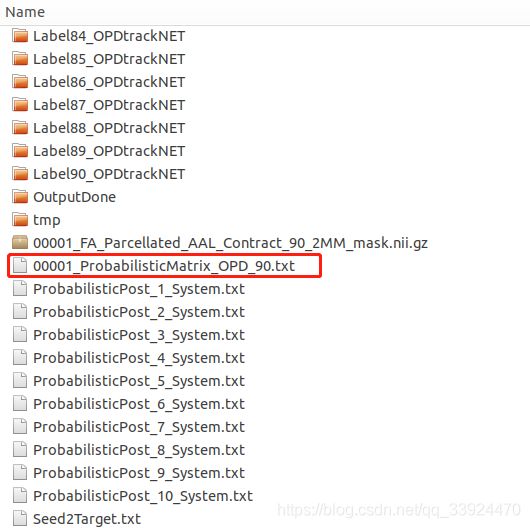
概率网络文件夹下面有很多文件,大多是计算过程中产生的中间结果,最后的概率网络结果如上图的红色框所示,要注意概率网络不是对称网络,因此在对概率网络进行分析之前,需要先对称化处理,即把网络中对称位置的值相加然后除以二。
Part 3 质量控制文件 quality_control

该文件夹存放的是FA图像、预处理后的T1、每一步的配准结果等等,以观察每一步的配准效果如何。
如第一个FA文件夹,打开后是一张三维显示的FA图像:

由于diffusion的图像不像fMRI图像可以通过头动参数等判断图像的质量如何,因此只能根据这种FA图像来进行初判。如观察灰质白质的对比度是否合适,白质的轮廓是否清晰等等。
下图是FA图像到标准空间的配准结果,红色的是标准空间的图像轮廓,下图展示的是配准得比较好的结果:
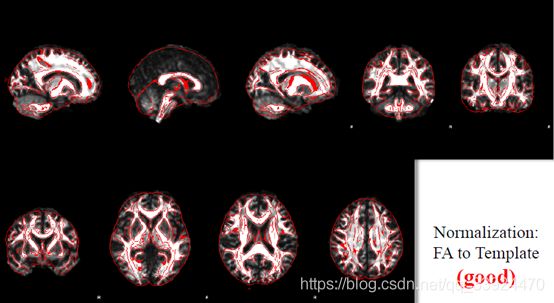 图片出自官方教程
图片出自官方教程
下图是T1图像配准到标准空间的结果,可以明显看出效果较差:
 图片出自官方教程
图片出自官方教程
通过观察这些配准结果,可以知道自己每一步的数据质量、数据配准、预处理等等是否有问题。
以上就是PANDA的所有笔记内容,PANDA运行的出错概率相对较低,个人觉得更重要的是要弄清楚每一步的输出结果代表什么,才可以更好地进行下一步的分析。
文中如有错误的地方,欢迎交流指出!