今天给大家带来的文献研究方向是有关于中国人群基因组组装以及全基因组分析[Whole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han Genome,IF=6.5+],各民族的区域参考基因组仍然是相关医学研究的短板,人口研究将在很大程度上依赖于目前所缺乏的中国北方参考基因组。为了揭示疾病和生理性状的遗传机制,在这里报告了CASPMI项目的初步成果,包括北方汉族参考基因组(NH1.0)的从头组装和来自中国大部分地区健康人的全基因组分析。
中国人群全基因组分析及北方汉族基因组的从头组装
方法
样本信息
共有597名受试者(246名男性和351名女性)参加了这项研究,北方血统和南方血统共识别出455个样本,分别为339个北方人(NH)和116个南方人(SH)。
基因组组装
PacBio平台的long reads和10x Genomics平台linked reads分别使用CANU和Supernova工具来组装。使用Illumina双端读长来校正PacBio的重叠群(contig)中的错配和小插入缺失(small indel)。为了合并两个已组装基因组的数据集,通过使用MUMMER将重叠群与从linked reads组装的长序列片段(scaffold)进行比对,来识别两个数据集之间的序列重叠,并使用Gapcloser将双端读长用来填充scaffold间的空隙。
SNV/indel calling
在去除测序接头和低质量碱基之后,使用BWA将读长映射到人类参考基因组(Hg19)。使用Picard工具去排序映射结果为BAM格式,并标记PCR扩增的重复,然后使用GATK进行变异召回和过滤。为验证SNV calling的准确性,随机抽取了890个SNV,对含有SNV位点的片段进行PCR扩增和Sanger测序验证。
结构变异的检测
为了识别3个中国个体基因组中的结构变异,用LASTZ将scaffold序列与GRCH38进行比对,并使用“SOAPsv”包获得候选的结构变异。然后,通过比较单端读长与配对读长的比率(S/P ratio)来筛选结构变异。对于CASPMI队列,以hg19作为参考,使用CREST,Pindel和Control-FREEC来检测结构变异,保留大于50bp的结构变异以供进一步分析。对于每个样本,将重叠超过80%的结构变异合并;在样本间,将重叠超过50%的结构变异合并以获得无冗余的结构变异。
基因组变异的注释
使用ANNOVAR和内置的数据库(RefGene、GWAS Catalog等)来注释基因组变异的等位基因频率和相关基因功能。使用Kobas进行变异相关基因的通路富集分析。计算启动子DHS信号与远端DHS信号的Pearson相关系数r,如果该系数大于0.7,则认为远端DHS与启动子DHS所在的基因相关。使用在线工具RegomeDB注释非编码SNP周围的DNA特征和调控元件。
关联分析
关联分析由PLINK工具完成。为了进行代谢性状关联分析,从EHR系统中收集了表中列出的10个表型,数量性状的关联检验采用PLINK线性回归模型,病例对照的关联检验采用Logistic回归模型。在全基因组关联分析中,选择群体频率大于0.05且单个样本中缺失基因型最多的SNP进行分析。
突变特征识别
对于来自所有个体的SNV calling,作者重点关注那些在整个样本中只出现一次的变体,也称为单例(singleton)。在每个个体中有大于10个读长支持且没有收录在dbSNP中的结构变异,被识别为新的单例。根据突变位点及其两侧碱基的不同,将每个新的单例划分成96个可能突变的三核苷酸,然后生成所有样本的三核苷酸计数矩阵。从hg19中提取每种突变类型的背景,然后利用R包“MutationalPatterns”来估算出符合COSMIC已知特征的突变谱。
结果
NH1.0基因组的组装
通过与Illumina双端读长比较,校正了PacBio重叠群的错配和小插入缺失,序列校正后组装的长度明显增加。在将PacBio重叠群与10x Genomics的scaffold进行比对和冗余筛选后,把两个基因组进行整合。然后利用BioNano光学图谱数据使scaffold N50提高到46.63Mb,共包含5574个scaffold,总组装长度为2.89 GB(下表),并将这个组装的基因组命名为NH1.0。
NH1.0的scaffold与GRCh38基因组的序列比对显示这两个基因组之间高度一致。与中国人现存的两个个体基因组(HX1和YH2.0)相比,NH1.0 有更长的scaffold,跨越了15条染色体臂,覆盖了85%以上的常染色体区域,并提高了染色体水平的完整性(下表)。NH1.0基因组完全填补了GRCh38上99个测序间隙,并识别了749个新序列。证据表明表明NH1.0基因组比参考基因组GRCh38更能代表中国人群。NH1.0还分别与HX1和YH2.0基因组重叠了39.0%和37.0%的SNV(图A)。相比之下,大多数结构变异都是南北个体特异性的(图B)。
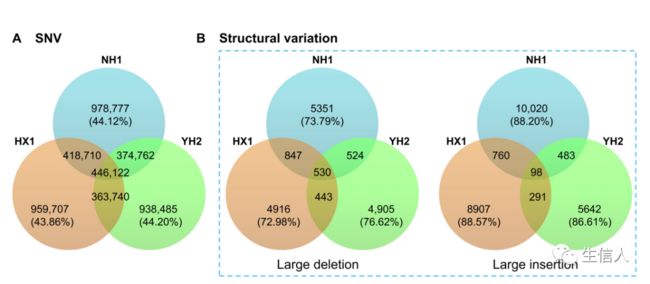
CASPMI队列中的测序和变异分析
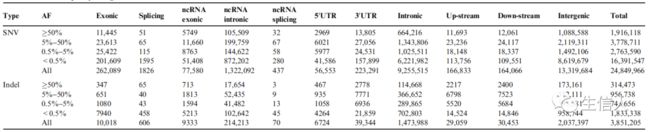
为了研究中国人群的遗传变异,作者对CASPMI队列的597名受试者进行了Illumina双端全基因组测序(WGS)。使用GATK识别出 SNV和小插入缺失,其中22.9%的SNV和33.0%的indel为常见类型,90.9%的变异位于基因间隔区和内含子区域。在线粒体中检测到1211个变异,其中897个(74.1%)位于基因区域。此外,部分非同义SNV和移码indel的等位基因频率大于5%(下表)。为了评估SNV calling的准确性,随机选择了890个变体进行Sanger测序,其中99.1%得到了验证。如图A所示,该队列显著地识别了大量dbSNP和1KGP中没有的SNV和indel,这些新变异将有助于中国和全球的人口和健康相关研究。
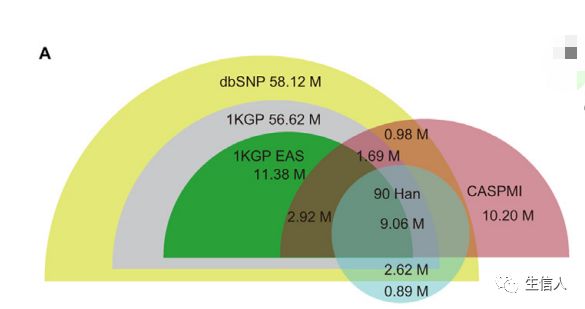
群体特异性SNP和代谢相关注释
为了进一步识别CASPMI队列的群体特异性变异,首先,比较队列样本和1KGP中不同群体的SNP和indel频率,使用南亚和欧洲等人基因组作为边远群体来分析人口变异的频率,通过筛选频率差异,将这组群体识别出的特异性变异命名为hfCAS-EAS,代表CASPMI队列中相对高频的SNP。接下来,对hfCAS-EAS数据集进行了KEGG通路富集分析,最显著的是代谢通路(图B)。然后,作者筛选了GWAS Catalog和hfCAS-EAS数据集中共有的40个SNP,这些重要的SNP所对应的38个基因与各种代谢相关的特征和疾病相关,包括腰围、BMI和脂代谢等。对其中17个SNP进行了包括腰围、空腹血糖水平和血压等表型数据的定量关联分析,结果发现KAT8中的rs1549293与男性的腰围显著相关,而NR2F2-AS1基因频率在人群中差异很大,在CASPMI队列和东亚人中很高,在hfCAS-EAS数据集中达到最高的92%,队列中的246名男性中,这个SNP与较大的腰围有显著关联(图E和表)。进一步证据表明,包含该SNP的增强子区域与CASPMI队列中北方男性较大的腰围相关,其中T等位基因与PU.1的结合较弱可能会改变肥胖相关基因FUS和HSD3B7的表达(图F,G)。


北方和南方群体的遗传分化
作者通过分析NH组和SH组变异的频率分布来研究汉族群体的遗传结构,根据455例有明显血统信息的常染色体SNP,使用VCFtools计算中国北方和南方汉族人群之间的FST(群体间遗传分化指数)。结果显示,有几个重要Fst信号峰聚集在一起,主要位于第6、11、14和19号染色体上(图A)。最高的Fst信号聚集在14号染色体上,靠近与免疫球蛋白位点(IGH)相关的基因。此外,与循环叶酸、维生素B12或脂质代谢相关的MTHFR、TCN2、FADS1和FADS2基因在北方人和南方人之间存在显著的遗传多样性(图B-D),表明在汉族人群进化过程中,体现了不同的环境暴露和生活方式的选择。

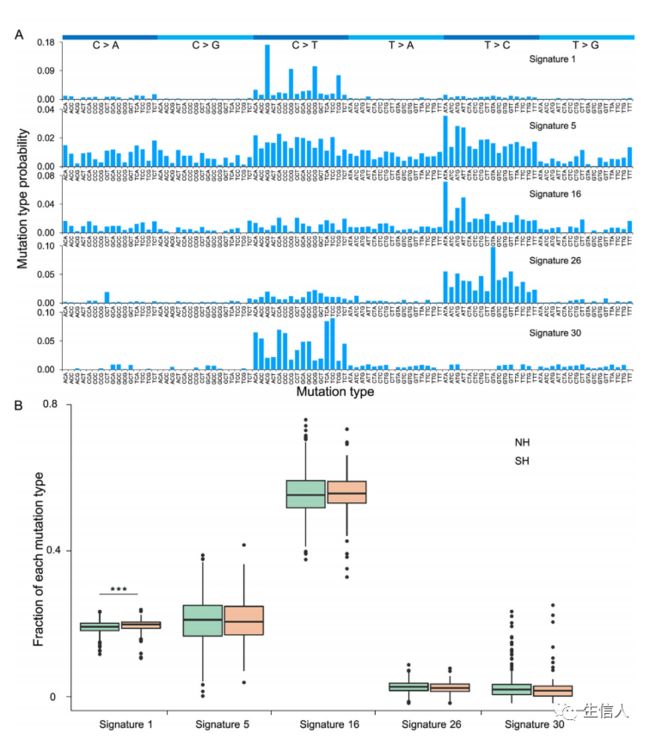
新单例的突变特征与种群分布
对于大量识别到只存在于CASPMI群体中个体SNV的新单例,作者分析它们的突变特征来探索可能驱动分离变异产生的潜在机制。首先,根据突变位点及其两侧的两个碱基将SNV分为96类。然后,采用非负矩阵分解(NMF)来识别特定的突变特征。如图A所示,在COSMIC中识别了代表不同突变过程的5个突变特征。其中,特征16(一种已证实在全球人群中分离并仅在COSMIC中的肝癌患者中发现的突变类型)显示出最高的个体负荷(图B)。在这5个突变特征中,特征1在两组之间呈现显著差异。特征1代表由胞嘧啶脱氨基引起的突变,这种类型的突变在南方汉族人群中的发生率较高,这意味着南方人可能有更长的世代时间。在个体水平上,特征5和16占很大比例,此外,特征5在不同个体之间呈现最大的差异。综上所述,北方和南方汉族之间拥有非常相似的突变谱,突变特征负荷的群体差异和个体差异可能与平均世代时间有关。
CASPMI队列中的结构变异
与dbVar和DGV相比,作者识别到了6万多个新的结构变异。在所有的结构变异中,97.8%为缺失,其中等位基因频率<0.05的占88.6%。此外,三分之二的结构变异的等位基因频率<0.005,表明大多数结构变异在测序人群中是罕见或非常罕见的。
结构变异多位于重复区,此外,散在重复序列比串联重复序列更易于被证实与大多数结构变异相关。通过对其平均长度的比较,还发现具有复杂重复元件的结构变异通常比其他结构变异更长。最后,为了研究结构变异的功能效应,将含有结构变异的基因定位到GWAS Catalog。如表所示,体重指数和肥胖相关性状是前10个显著富集的功能类,这与hfCAS-EAS数据集的KEGG富集分析结果相似。这揭示了本研究中确定的SNV和结构变异都与代谢相关性状有关,这可能与CASPMI队列中男性代谢综合征的高患病率有关。
