基于上游分析获得表达矩阵后,就可以进行差异分析了,最基础的莫过于利用DEseq2包寻找差异基因。学习自b站视频生信技能树转录组视频~
0、数据准备及前处理
- 由于目前在家不方便获得数据,因此参照教学视频使用airway包的现成数据。
如链接中官方介绍,airway包背景是使用地塞米松处理的四个人气道(支气管?)平滑肌细胞(即共8个数据)进行rna-seq实验,分析得到的基因表达矩阵。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("airway")
library(airway)
data(airway)
airway
class(airway)
- 如上返回结果,airway为RangedSummarizedExperiment类型的对象,是将数据归档R包的一个好方法,使用时主要会涉及到两个函数操作
assay(data)表示取观测为基因名,变量为样本信息的表达矩阵;
colData(data)则是取记录详细的样本信息;
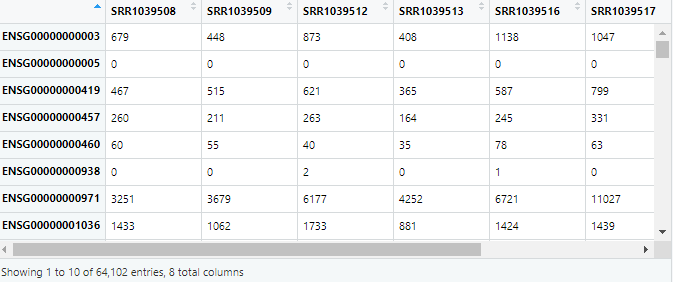
exp=assay(airway) #取原始表达矩阵数据
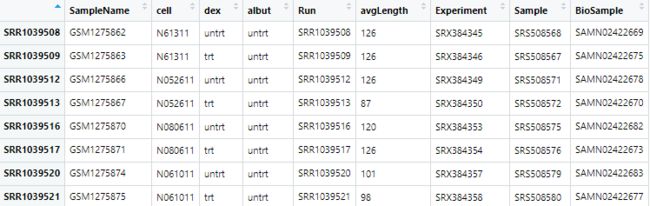
tmp=colData(airway)
tmp=as.data.frame(airway)
#由于后续分析需要分组信息,因此这里我们取下第三列信息
group=colData(airway)[,3]
class(group)
值得注意的是
colData(airway)得到的结果并非直接为真正的表格,还需要用as.data.frame()函数转换下,后面分析还会遇到类似的问题。
index1=c(rowSums(exp)>0)
class(index1)
exp2=exp[index1,]
- 此处操作的目的在于将没有read比对上的基因行删去,简洁数据~
1、DEseq2包差异分析
(1)设置单列分组样本信息表
group
sample=c(colnames(exp2)) #取样本名信息
f=as.data.frame(group)
rownames(f)=c(sample) #设置行名为样本名信息
colnames(f)='condition' #列名

(2)DESeq2分析
library("DESeq2")
dds=DESeqDataSetFromMatrix(countData = exp2,
colData = f,
design = ~ condition)
dep=DESeq(dds)
res=results(dep,contrast=c("condition","trt","untrt"))
这里要注意一下,所谓差异分析,即比较实验组与对照组相比,基因表达变化情况。通过
contrast=参数分别设置样本分组信息表的列名,实验组名(numerator,分子),对照组名(denominator,分母);即研究分子/分母的结果
DEG1=as.data.frame(res)
write.csv(x=DEG1,file="res.csv")
- 返回的六列值中重点关注 log2FoldChange,pvalue,padj三列内容。
log2FoldChange中的FoldChange即倍数变化的意思。某一基因表达 实验组/对照组,可分为两种结果:大于1,即上调;小于1,即下调。利用log函数将其分别转化成正数、负数,更便于理解。(通常为了防止取log2时产生NA,我们会给表达值加1(或者一个极小的数),也就是log2(B+1) - log2(A+1));
pvalue是假设检验的p值,值越小(一般标准0.05)即说明log2FoldChange数据的可靠性越强;padj为adjusted p-value值,也称Q-value、FDR(false discovery rate)为效验过后的p-value值,更有信服力。
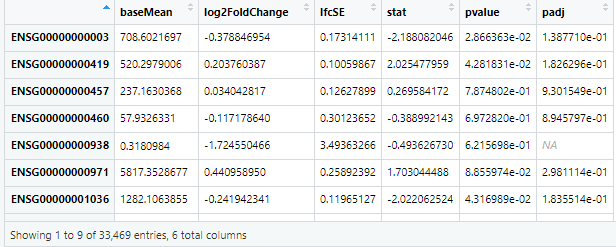
resOrdered=res[order(res$pvalue),] #order函数是给出从小到大排序后的位置(默认升序)
sum(res$padj<0.1, na.rm=TRUE) # na.rm 为remove na值,否则会影响统计量
sum(res$pvalue<0.05, na.rm=TRUE)
sum(!is.na(res$pvalue))
resSig=subset(resOrdered, padj<0.1)
DEG2=as.data.frame(resSig)
write.csv(x=resSig, file = "results.csv")

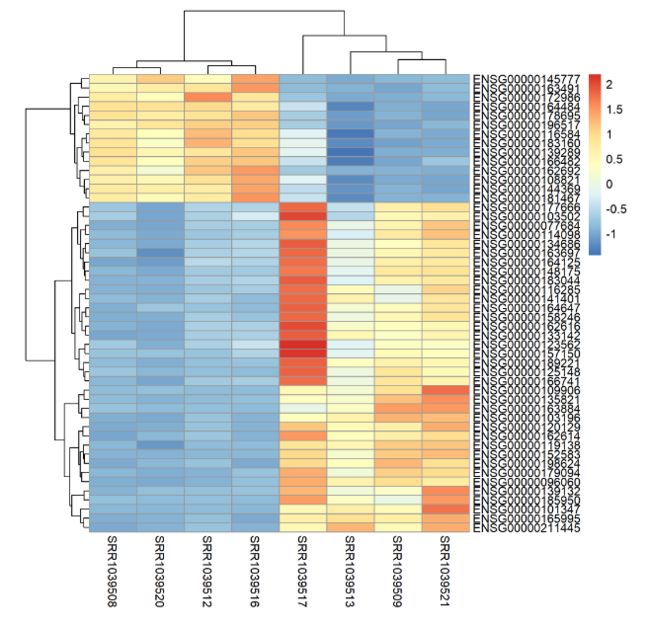
2、绘制热图
- 根据基因表达矩阵比对上的read数目绘制
- 这里选取最有意义的50个基因比对read数目为例
library(pheatmap)
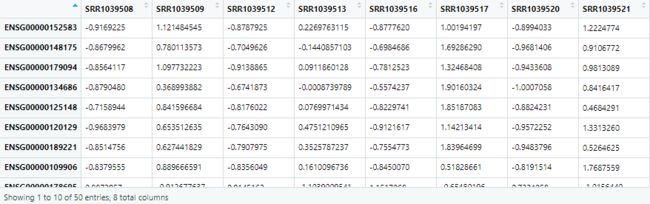
choose_gene=head(rownames(DEG2),50)
#根据DEG2结果,提取目标基因名
choose_matrix=exp2[choose_gene,]
choose_matrix=t(scale(t(choose_matrix)))
一般绘制热图,都需要对数据进行归一化处理。这里用到了
t()转置函数的原因是因为基因表达矩阵表中观测为基因,变量为样本。而scale()归一化函数是针对每一列单独处理的。因为每一个基因的本身情况各不相同,而且我们的目的是基于实验组与对照组的比较来研究基因表达变化,所以需要转置下表达矩阵(观测为样本,变量为基因),然后再进行对每一列的归一化处理;最后再进行一次转置,即得到下图结果。
pheatmap(choose_matrix,filename = 'DEG_top50_heatmap.png')
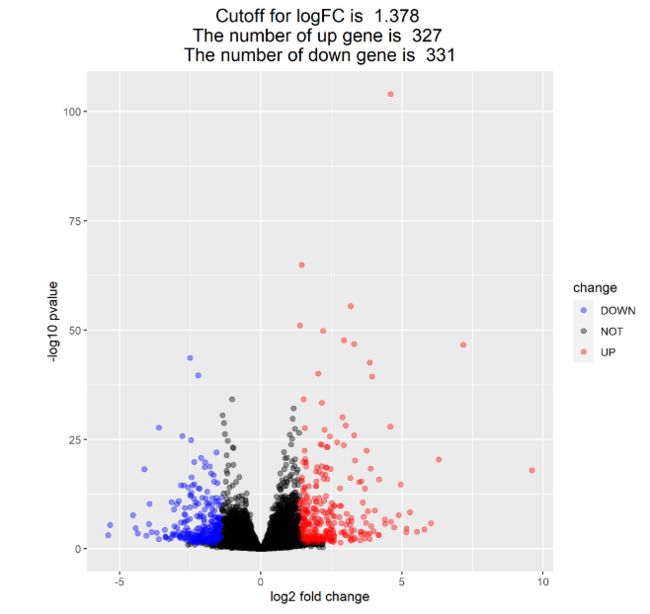
3、绘制火山图
- 本质上就是根据DEG数据里基因的log2FoldChange与pvalue绘制点图
- 把点分成三类:上调基因、下调基因与无显著改变基因。
DEG1=na.omit(DEG1)
(1) 设定分类判断阈值并增加分类列
logFC_cutoff=with(DEG1,mean(abs(log2FoldChange))+2*sd(abs(log2FoldChange)))
logFC_cutoff
2^logFC_cutoff
# 返回结果为 3.67
DEG1$change=as.factor(ifelse(DEG1$pvalue<0.05 & abs(DEG1$log2FoldChange)>logFC_cutoff,
ifelse(DEG1$log2FoldChange>logFC_cutoff,'UP','DOWN'),'NOT'))
(2)编写图中的注释信息
this_tile=paste('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG1[DEG1$change=='UP',]),
'\nThe number of down gene is ',nrow(DEG1[DEG1$change=='DOWN',]))
- 其中
\n表示换行符
(3)利用ggplot2绘制火山图
library(ggplot2)
g=ggplot(data=DEG1,
aes(x=log2FoldChange,y=-log10(pvalue), #这里将pvalue取负对数
color=change)) +
geom_point(alpha=0.4,size=1.75) + #绘制点图
theme_set(theme_set(theme_bw(base_size=20))) +
xlab("log2 fold change")+ylab("-log10 pvalue") + #轴标签
ggtitle(this_tile)+theme(plot.title=element_text(size=15,hjust=0.5)) +
scale_color_manual(values=c('blue','black','red')) #设定颜色
ggsave(g,filename='volcano.png')
# 如下图,一般会重点关注两边的,偏上方的基因点
以上是基于基因表达矩阵进行差异分析以及绘制热图、火山图的大致流程。这应该下有分析简单的开始,后期还有富集分析等,再慢慢学习吧~