引言:前面几期中,我们学习了如何下载TCGA数据、预处理和差异分析,那么今天我们继续来看看如何将利用差异分析的结果绘制热图和火山图。TCGAbiolinks包的功能太强了,几乎可以实现TCGA数据一站式分析,故今天小编仍然用TCGAbiolinks包中的函数完成今天的演示。
若有疑问,可关注文末公众号,回复“R语言实战”加入讨论群。
一、加载预处理文件
setwd("---") #记得设置正确的设置默认路径
library("TCGAbiolinks") #加载TCGAbiolonks包
##1 加载前几期的数据,获取45配对的肿瘤样本和正常组织样本
TCGA_LIHC_data <- read.csv(file = "TCGA_LIHC_final.csv",
header = T,
row.names = 1,
check.names = FALSE #保证列名不发生自动更正)
#获取正常组织与肿瘤组织的barcodes
samplesNT <- TCGAquery_SampleTypes(colnames(TCGA_LIHC_data), typesample = c("NT"))
samplesTP <- TCGAquery_SampleTypes(colnames(TCGA_LIHC_data), typesample = c("TP"))
#获取配对正常组织与肿瘤组织的barcodes
paired <- intersect(substr(samplesNT,1,12),substr(samplesTP,1,12))
length(paired)
#[1] 45
#用前12位barcodes为桥梁,建立肿瘤与正常样本barcodes的数据框
NT <- data.frame(NT1=substr(samplesNT,1,12),NT2=samplesNT)
TP <- data.frame(TP1 =substr(samplesTP,1,12),TP2=samplesTP)
TP_NT <- merge(TP,NT,by.x = "TP1",by.y = "NT1")
head(TP_NT,3)
# TP1 TP2 NT2
# 1 TCGA-BC-A10Q TCGA-BC-A10Q-01A-11R-A131-07 TCGA-BC-A10Q-11A-11R-A131-07
# 2 TCGA-BC-A10R TCGA-BC-A10R-01A-11R-A131-07 TCGA-BC-A10R-11A-11R-A131-07
# 3 TCGA-BC-A10T TCGA-BC-A10T-01A-11R-A131-07 TCGA-BC-A10T-11A-11R-A131-07
#1.1获取配对正常组织的barcodes:
TP <- TP_NT$TP2
#1.2获取配对肿瘤组织的barcodes:
NT <- TP_NT$NT2
二、配对肿瘤组织与正常组织数据的下载与预处理
#2 参照前面几期进行数据下载和数据预处理:
queryDown <- GDCquery(project = "TCGA-LIHC",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = c(TP, NT))
GDCdownload(queryDown,method = "api", directory = "GDCdata",
files.per.chunk = 10)
#2.1 将数据准备成R语言可处理的形式
dataPrep1 <- GDCprepare(query = queryDown, save = TRUE, save.filename =
"LIHC_case2.rda")
#2.2数据预处理:根据样本与样本之间的spearman相关系数去掉离群值
dataPrep2 <- TCGAanalyze_Preprocessing(object = dataPrep1,
cor.cut = 0.6,
datatype = "HTSeq - Counts")
#2.3选择肿瘤纯度大于60%的肿瘤样本(因为这是基于前面两期获得的肿瘤样本barcodes,故这里获得的肿瘤样本的肿瘤纯度均大于60%,可不执行此步骤)
# purityDATA <- TCGAtumor_purity(colnames(dataPrep1), 0, 0, 0, 0, 0.6)
# # filtered 为被过滤的数据, pure_barcodes是我们要的肿瘤数据
# Purity.LIHC<-purityDATA$pure_barcodes
# normal.LIHC<-purityDATA$filtered
#
# #获取肿瘤纯度大于60%的样本+正常组织样本,共计90个样本
# puried_data <-dataPrep2[,c(Purity.LIHC,normal.LIHC)]
# #有45个肿瘤纯度大于60%的样本
#2.4基因注释
library("SummarizedExperiment")
rowData(dataPrep1)
# DataFrame with 56512 rows and 3 columns
# ensembl_gene_id external_gene_name original_ensembl_gene_id
#
# ENSG00000000003 ENSG00000000003 TSPAN6 ENSG00000000003.13
# ENSG00000000005 ENSG00000000005 TNMD ENSG00000000005.5
# ENSG00000000419 ENSG00000000419 DPM1 ENSG00000000419.11
# ENSG00000000457 ENSG00000000457 SCYL3 ENSG00000000457.12
#将结果写入文件“puried.LIHC.cancer.csv”
rownames(puried_data)<-rowData(dataPrep1)$external_gene_name
write.csv(puried_data,file = "paried_puried.LIHC.csv",quote = FALSE)
#2.5使用EDAseq进行文库大小和GC丰度标准化
dataNorm <- TCGAanalyze_Normalization(tabDF = puried_data,
geneInfo = geneInfo,
method = "gcContent")
#2.6过滤低count的基因,并将结果输出
dataFilt <- TCGAanalyze_Filtering(tabDF = dataNorm,
method = "quantile",
qnt.cut = 0.25)
str(dataFilt)
#num [1:13082, 1:90] 11 5376 10551 610 1048 ...
#- attr(*, "dimnames")=List of 2
#..$ : chr [1:13082] "A1BG" "A1CF" "A2M" "A4GALT" ...
#..$ : chr [1:90] "TCGA-BC-A10Q-01A-11R-A131-07" "TCGA-BC-A10R-01A-11R-A131-07" "TCGA-BC-A10T-01A-11R-A131-07" "TCGA-BC-A10U-01A-11R-A131-07" ...
write.csv(dataFilt,file = "paired_TCGA_LIHC_final.csv",quote = FALSE)
#保留的是90个样本(前45肿瘤,后45正常组织)
三、差异表达分析
##3 差异表达分析:(前)45个肿瘤样本 vs (后)45个正常组织样本
TCGA_LIHC_data <- read.csv(file = "paired_TCGA_LIHC_final.csv",header = T,row.names = 1,check.names = FALSE)
mat1 <- TCGA_LIHC_data[,1:45]
mat2 <- TCGA_LIHC_data[,46:90]
DEG.LIHC.edgeR <- TCGAanalyze_DEA(mat1 = mat1,
mat2 = mat2,
pipeline="edgeR",
batch.factors = c("TSS"),
Cond1type = "tumor",
Cond2type = "normal",
voom =FALSE, ##设置了paired时,会出错(paired = TRUE),故此处未设置
method = "glmLRT",
contrast.formula = "Mycontrast =tumor -normal",
fdr.cut = 0.01, #设置过滤参数1,保留FDR<0.01的基因
logFC.cut = 1 #设置过滤参数2,保留logFC>1的基因
)
write.csv(DEG.LIHC.edgeR1,file = "paired_DEG_by_edgeR.csv")
四、TCGAanalyze_LevelTab() 增加不同分组条件下的gene平均表达量
应用:将差异表达基因在正常和肿瘤组织中的表达量数据添加到差异表达分析结果中。
主要用法:
TCGAanalyze_LevelTab(FC_FDR_table_mRNA, typeCond1, typeCond2, TableCond1,
TableCond2, typeOrder = TRUE)

image.png
R中具体示例:
#4.1 TCGAquery_SampleTypes()用于获取特定组织对应的barcodes,如肿瘤组织(TP)、正常组织(NT)
#设置typesample =NT,获取正常组织对应的barcodes
samplesNT <- TCGAquery_SampleTypes(colnames(dataFilt.LIHC.final), typesample = c("NT"))
#设置typesample=TP,获取肿瘤组织对应的barcodes
samplesTP <- TCGAquery_SampleTypes(colnames(dataFilt.LIHC.final), typesample = c("TP"))
dataDEGsFilt <- DEG.LIHC.edgeR[abs(DEG.LIHC.edgeR$logFC) >= 1,]
str(dataDEGsFilt)
# 'data.frame': 2647 obs. of 5 variables:
# $ logFC : num 1.23 -1.32 -1.12 2.17 -1.31 ...
# $ logCPM: num 7.1 2.52 3.33 4.87 1.32 ...
# $ LR : num 21.7 46.7 41.4 56.2 21.4 ...
# $ PValue: num 3.23e-06 8.14e-12 1.27e-10 6.70e-14 3.79e-06 ...
# $ FDR : num 1.04e-05 6.50e-11 8.39e-10 7.32e-13 1.21e-05 ...
#获取肿瘤组织对应的表达矩阵
dataTP <- dataFilt.LIHC.final[,samplesTP]
#获取正常组织对应的表达矩阵
dataTN <- dataFilt.LIHC.final[,samplesNT]
#4.2 将需要的参数传入dataDEGsFiltLevel ()函数中
dataDEGsFiltLevel <- TCGAanalyze_LevelTab(FC_FDR_table_mRNA=dataDEGsFilt,
typeCond1="Normal",
typeCond2="Tumor",
TableCond1=dataTN,
TableCond2=dataTP)
#4.3 查看结果
head(dataDEGsFiltLevel,2)
# mRNA logFC FDR Normal Tumor Delta
#HP HP 1.184681 7.568768e-06 1087091.5 490505.1 1287856.1
#APOA2 APOA2 -1.155957 6.493243e-06 308058.6 707503.1 356102.5
差异表达分析的结果如下:
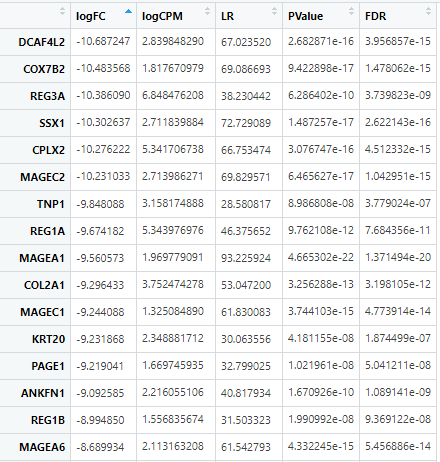
image
添加两种条件下基因的平均表达水平后的结果如下:

image
五、TCGAvisualize_PCA()实现主成分分析
主要用法:
TCGAvisualize_PCA(dataFilt, dataDEGsFiltLevel, ntopgenes, group1, group2)
参数详解:

R中具体示例:
#由于在TCGAanalyze_LevelTab()中,我们已经得到了一些参数,故可将参数直接带入主成分分析的函数中。
pca <- TCGAvisualize_PCA(dataFilt= dataFilt.LIHC.final,
dataDEGsFiltLevel=dataDEGsFiltLevel,
ntopgenes = 100,
group1=samplesNT,
group2=samplesTP
)
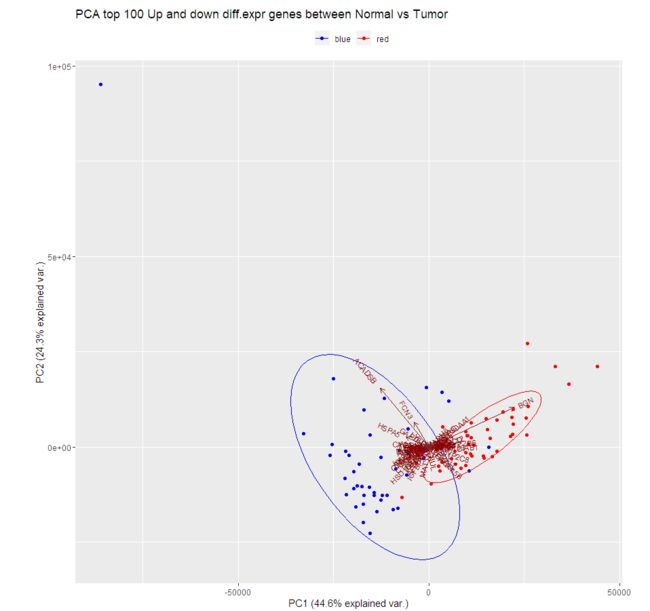
image
六、绘制差异表达基因的热图
主要用法:等号后面对应的参数为TCGAvisualize_Heatmap()的默认参数。
TCGAvisualize_Heatmap(data, col.metadata, row.metadata,
col.colors = NULL, row.colors = NULL, show_column_names = FALSE,
show_row_names = FALSE, cluster_rows = FALSE,
cluster_columns = FALSE, sortCol, extrems = NULL,
rownames.size = 12, title = NULL, color.levels = NULL,
values.label = NULL, filename = "heatmap.pdf", width = 10,
height = 10, type = "expression", scale = "none",
heatmap.legend.color.bar = "continuous")
参数详解:
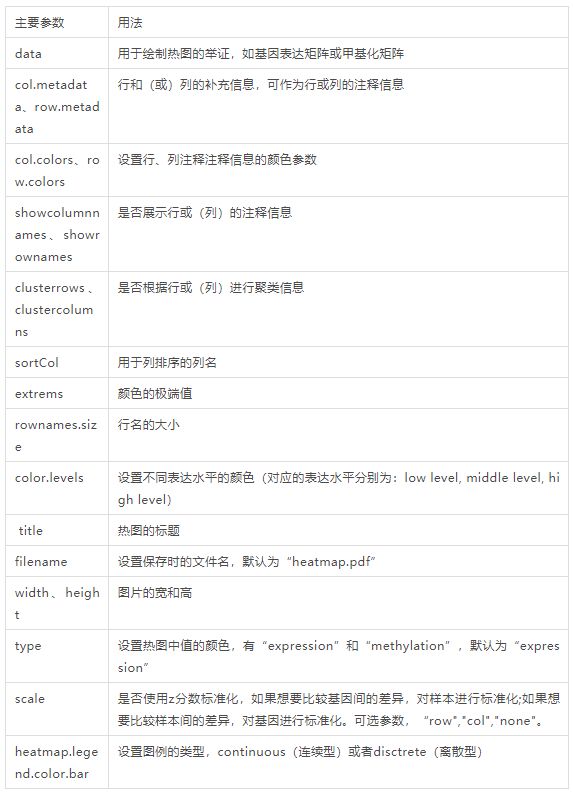
R中具体示例:
#6.1 获取差异表达基因的表达水平
datDEGs <- dataFilt.LIHC.final[match(rownames(DEG.LIHC.edgeR),rownames(dataFilt.LIHC.final)),]
str(datDEGs)
#'data.frame': 2647 obs. of 90 variables:
#前45是肿瘤样本,后45是正常组织样本
#6.2 根据临床信息构造patient的metadata信息;或(和)基因的相关信息(此处不添加基因的注释信息)
#获取每一个患者barcode(barcode的前12位代表的patient信息,前16位代表的是sample信息)对应的临床信息,但是其barcodes与datDEGs_test顺序不一致
query <- GDCquery(project = "TCGA-LIHC",
data.category = "Clinical",
file.type = "xml",
barcode = substr(colnames(datDEGs),1,12))
GDCdownload(query)
clinical <- GDCprepare_clinic(query,"patient") #48个样本
##根据表达矩阵中的样本barcodes对样本临床信息匹配
datDEGs_test_barcodes <- as.data.frame(substr(colnames(datDEGs),1,12), ncol=1)
colnames(datDEGs_test_barcodes) <-"LIHC_patient_barcode"
m <- clinical[match(datDEGs_test_barcodes[,1], clinical[ , 1 ]),]
str(m)
#'data.frame': 90 obs. of 82 variables:
table(duplicated(m))
#FALSE TRUE
#45 45
#使用table(duplicated())查看m矩阵中是否有重复数据。
#这里的重复数据来源(肿瘤组合和癌旁正常组织来源于同一患者)
##由于使用的是配对正常样本和肿瘤组织,其对应的患者12位barcodes是一致的,在使用TCGAbiolinks包自带的热图绘制函数时会出现样本信息匹配错误,故小编在这里使用pheatmap()绘制热图。
```r
library(pheatmap)
#原始做图结果
pheatmap::pheatmap(datDEGs,scale = "row",show_rownames = F,show_colnames = F)
上图为未设置其他参数时的原始如图结果,为增加图片的信息,可增加metadata信息(即行注释和列注释信息),注意pheatmap()的注释信息的列名必须与表达矩阵的行名、列名一致,演示如下:
#增加metadata信息
col.mdat <- data.frame(Sex=m$gender,
status=m$vital_status,
group=c(rep("tumor",45),rep("normal",45)))
rownames(col.mdat) <- colnames(datDEGs) #保证列注释信息的行名与样本名(对应列)一致
#设置图例的范围
bk <- c(seq(-1,6,by=0.01))
#绘制热图
pheatmap::pheatmap(datDEGs,scale = "row",show_rownames = F,show_colnames = F,
annotation_col = col.mdat3,
border_color=NA,
main = "Heatmap by pheatmap(edgeR)",
filename = "Heatmap_by_pheatmap.pdf",
color =c(colorRampPalette(colors = c("blue","white"))(length(bk)/2),
colorRampPalette(colors = c("white","red"))(length(bk)/2)) #设置图例的颜色,
legend_breaks=seq(-1,6,2),
breaks=bk )
以上为热图的输出结果,我们可以看到按照行(样本)进行聚类,基本上能够把肿瘤组织与正常组织分类开,说明两种组织的基因表达是具有差异的。相反,在不同存活状态和性别中,暂时未能发现与基因差异表达的相关性。
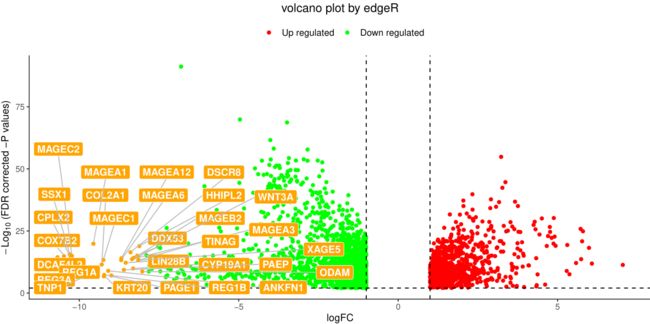
七、绘制差异差异表达分析结果的火山图
主要用法:
TCGAVisualize_volcano(x, y, filename = "volcano.pdf",
ylab = expression(paste(-Log[10], " (FDR corrected -P values)")),
xlab = NULL, title = "Volcano plot", legend = NULL, label = NULL,
xlim = NULL, ylim = NULL, color = c("black", "red", "green"),
names = NULL, names.fill = TRUE, show.names = "significant",
x.cut = 0, y.cut = 0.01, height = 5, width = 10,
highlight = NULL, highlight.color = "orange", names.size = 4,
dpi = 300)
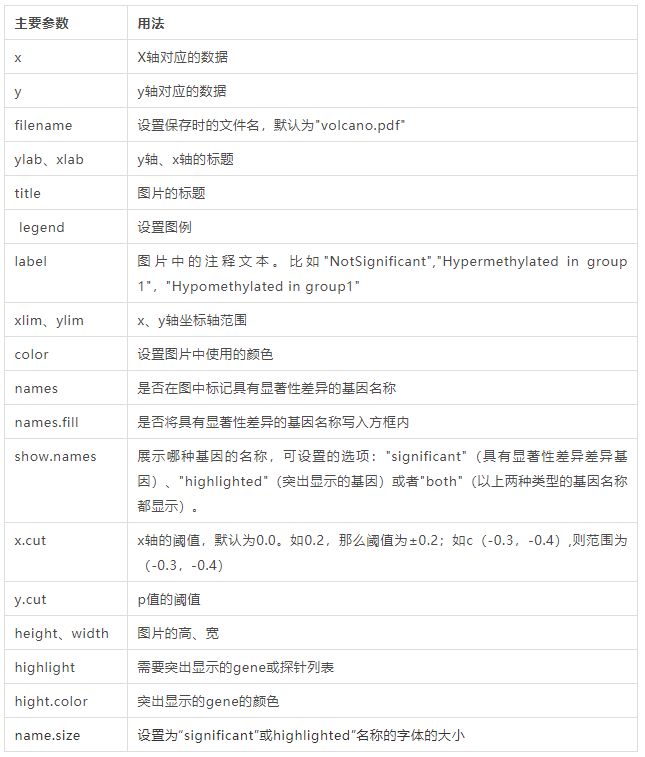
r中具体示例:
#为了做图的需要,突出显示FC≥8的gene名称
DEG.LIHC.filt<-DEG.LIHC.edgeR[which(abs(DEG.LIHC.edgeR$logFC) >= 8), ]
str(DEG.LIHC.filt)
#'data.frame': 29 obs. of 5 variables:
#共有29个基因满足|logFC|≥8
TCGAVisualize_volcano(DEG.LIHC.edgeR$logFC,
DEG.LIHC.edgeR$FDR,
filename = "TumorvsNormal_FC8.edgeR.pdf",
xlab = "logFC",
names = rownames(DEG.LIHC.edgeR),
show.names = "highlighted",
x.cut = 1,
y.cut = 0.01,
highlight = rownames(DEG.LIHC.edgeR)[which(abs(DEG.LIHC.edgeR$logFC) >= 8)],
highlight.color = "orange",
title = "volcano plot by edgeR")
title = "volcano plot by limma")
如下为此次分析的火山图结果,通过查看图片,可以发现一些基因在肿瘤组织中表达量升高较高,而一些基因的肿瘤组织的表达量低于正常组织中,具体它有什么含义,就需要查阅文献明确。
image
八、结语
今天的热图和火山图就暂告一段落。但在实际过程中应该结合自己的数据,调整一些参数和分组,以得出更有意义的结论,为科研助力......接下来我们将使用TCGAbiolinks包继续演示TCGA数据中甲基化分析,我们一起努力哦~~~
