B站视频地址:https://www.bilibili.com/video/av49363776?from=search&seid=17709076368945641839
特此致谢:生信技能树
关于数据下载,其实有好多种方法,有firhose, 有XENA,甚至还有专门的R语言包比如TCGAbiolinks等等。当然GDC-client是官方软件下法,所以,我就选择了官方下载。这个在发表文章的时候应该可信度更高,也更为可靠。
当然,我也差点被这一章逼疯。。。毕竟我前前后后看了不下10次,就差一个一个字听了。甚至一度出现了:字我都认识,连起来我就不懂的情况。
1.数据下载
首先,进入TCGA的官方网站,现在全部和GDC整合在了一起了。https://portal.gdc.cancer.gov/

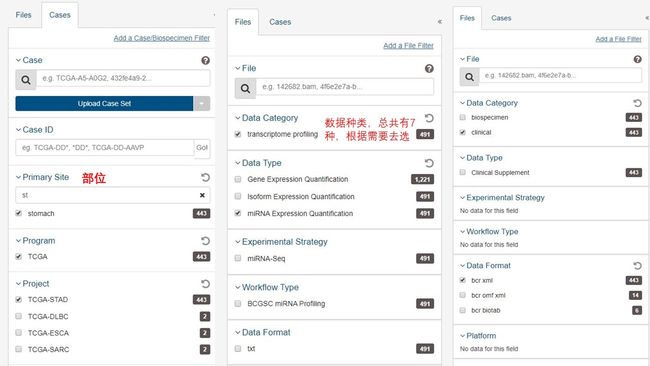
点击蓝色方框:Repository
mirna的数据下载:见下图第一、二部分,选择好之后点击mainifest进行mainnifest的下载
clinical的下载:参见下图第三部分,同样是下载mainifest
此外!clinical一定一定记得下载他的 json,这个将在后面发挥非常大的作用。(鬼知道当时我经历了什么,差点要疯)
进入gdc-client的官方网站: https://gdc.cancer.gov/access-data/gdc-data-transfer-tool选择合适的版本进行下载,解压然后提前建好一个空文档用来准备装数据的
修改环境变量:控制面板--系统---高级设置---环境变量---path--编辑--新建(找到解压好gdc-client的路径添加进去)这样就可以直接调用,避免后面报错。然后,我发现,电脑终端是不是都有点像,Linux的终端还五彩斑斓一点。Linux的界面也好好看!
C:\Users\Lenovo>G:
G:\>cd mirna
G:\mirna>gdc-client.exe download -m G:\mirna\STAD-mirna.txt
(这个TXT就是之前的mianifest,直接把mainnifest往DOS窗口一拖,他就会自动显示路径了)
然后就是漫长的下载,ヽ( ̄︿ ̄ )—C<(/;◇;)/ 我下载了一天多,中间经历了突然断网,也没记得设置断点续传(能设置这个吗,我就当能吧。我下载数据的经历告诉我,半夜三四点的网速特别好。)下载了mianifest之后,记得改名字,不然分不清哪个是哪个。
提取临床信息
rm(list=ls())
options(stringsAsFactors = F)
#######Load the packages required to read XML files.
library("XML")
library("methods")
dir='G:/mirna/clinical' ##设置临床信息所在的路径
all_fiels=list.files(path = dir,pattern='*.xml$',recursive=T)
###
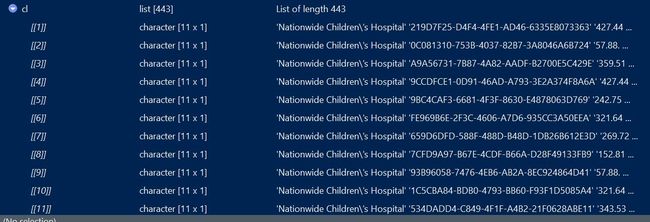
cl = lapply(all_fiels
, function(x){
result <- xmlParse(file = file.path(dir,x))
rootnode <- xmlRoot(result)
xmldataframe <- xmlToDataFrame(rootnode[2])
return(t(xmldataframe))
})
###关于rootnode[1]还是rootnote[2]还是有区别的,参见下图,rootnode[1]提取的不是临床信息
cl_df <- t(do.call(cbind,cl))
save(cl_df,file = 'STAD-clinical.Rdata')
提取表达矩阵
https://www.jianshu.com/p/af9b257c8a20
首先,谢谢小洁老师的教程,看着教程然后解决了一个难题
PS:我也不敢保证我接下来代码全对,因为这里当时真的非常混乱
###我是分开提取的,现在提取表达矩阵
rm(list=ls())
options(stringsAsFactors = F)
meta <- jsonlite::fromJSON("metadata.cart.2020-02-01.json")
###看到这个json文件了吗
entity <- meta$associated_entities
meta$associated_entities[[1]]
jh = function(x){as.character(x[4])}
###一定要记得看一下自己数据的submitter-id在第几列,有时候在第三列,有时候在第四列。
jh(entity[[1]])
###"TCGA-BR-8367-01A-11R-2343-13"
mis = dir("G:/mirna/micro-rna",pattern = "*mirnas.quantification.txt$",recursive = T)
ex = function(x){
result <- read.table(file.path("G:/mirna/micro-rna",x),sep = "\t",header = T)[,1:2]
return(result)}
head(mis)
ID = sapply(entity,jh)
file2id = data.frame(file_name = meta$file_name, ID = ID)
mis2 = stringr::str_split(mis,"/",simplify = T)[,2]
mis2[1] %in% file2id$file_name
head(match(mis2,file2id$file_name))
row_tcga = file2id[match(mis2,file2id$file_name),]
rownames(mi_df) = row_tcga$ID
mi_df[1:4,1:4]
吊诡的地方来了

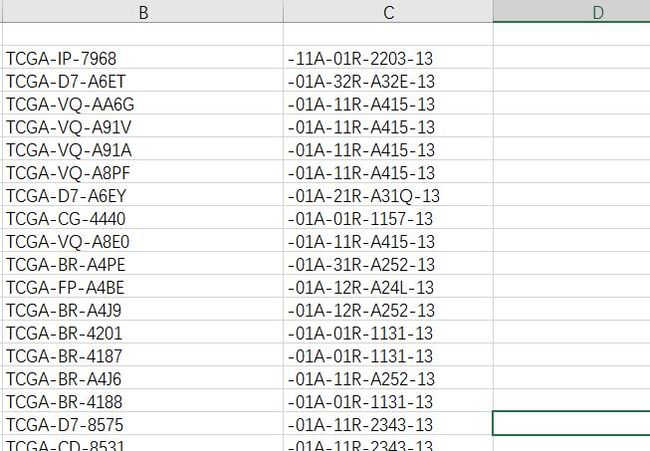
因为我一直想把表达矩阵和临床信息一起合并了,方便后面分析啥的。然而,问我打开两个数据框,表达矩阵和临床信息的ID不同。。。一个长,一个短,但是二者前10个字母相同的。既然都打开了excel,那么就在excel解决嘛!我知道可以用R,但是我想偷偷懒啊。

效果如下:当然是保留前面那一列,后面不用我说了吧,用merge函数就完事儿了
rm(list=ls())
options(stringsAsFactors = F)
midf<-read.csv(file = "G:/mirna/process/mi_df.csv")
mirnacl<-read.csv(file = "G:/mirna/process/mirna-clinical.csv")
mirnaN<-merge(midf,mirnacl,by.x="bcr_patient_barcode",by.y="bcr_patient_barcode")
write.csv(mirnaN,file = "G:/mirna/process/mirnaN.csv")
至此,GDC的下载和整理就结束了,再往下就是下游分析了。
难是有点难,不过,陆陆续续也做了十来个小时。
最后,外面肺炎疫情严重,各位还是在家好好学习吧,记得做好防护。也希望一线的同行们平安归来。