今天跟大家分享的是十二月份发表在EBioMedicine杂志(IF:5.736)上的一篇文章Transcriptional network modulated by the prognostic signature transcription factors and their long noncoding RNA partners in primary prostate cancer,该工作首先基于TCGA基因表达数据识别原发前列腺癌组织和正常组织的差异表达基因,然后使用多元线性回归来评估转录因子的活性,并识别在前列腺癌中活性显著上调和下调的转录因子。评估了转录因子活性与临床病理学特征的相关性并建立16个转录因子的预后模型。然后开发了一个识别lncRNAs与转录因子相互作用的方法,识别特征转录因子-lncRNA互作,并构建数据库LNCTRN。
TF相关文章:
5+TF-lncRNA肿瘤调控网络构建
发文很难吗?你只需要一个逆向思维:肿瘤TF预后
意向分析(http://gaptechsxr.mikecrm.com/1vdMmqy)

在原发性前列腺癌中由预后转录因子及其长链非编码RNA共同调控的转录网络
一、摘要
转录调节在前列腺癌的发生和发展中起着重要作用。然而,阐明其基本的控制环路和机制需要付出相当大的努力。该工作对原发性前列腺癌的基因组、转录组和临床病理特征以及转录因子结合特征进行了综合分析,包括评估转录因子活性,识别具有预后价值的转录因子,以及识别长非编码RNA的顺式和反式调控。RNA免疫沉淀定量PCR验证了转录因子和长链非编码RNA之间的相互作用。使用RNA干扰分析探索所选的转录调节的作用。最终识别到作为预后特征的16个转录因子。然后进一步发现候选长非编码RNA与预后特征转录因子的相互作用,并通过随机方法检验其相互作用。进一步验证了MYC及其长链非编码RNA伙伴AL590617.2在候选靶点上的转录调控作用。此外,由转录因子及其相互作用的长链非编码RNA共同调控的调控网络图示在LNCTRN数据库(https://navy.shinyapps.io/lnctrn)中。具有预后特征的转录因子及其相互作用的长链非编码RNA可能是前列腺癌的有前景的生物标志物和/或治疗靶点。该研究提出的计算方法可用于探索其他类型癌症的关键转录调控因子。
二、数据及方法
1. TCGA数据及预处理
从GDC data portal下载TCGA的498例原发性前列腺癌肿瘤和52例正常组织的拷贝数变异、基因突变、基因表达和临床病理学数据。保留在超过10%的样本中counts per million values大于0.5的20433个基因。基因表达FPKM值进行log2转换,然后进行Z-score 标化(zFPKM)。使用 edgeR(fold change > 1.5,FDR< 0.05)识别差异表达基因5946个。通过TCGAbiolinks获得分子亚型、术前PSA水平和肿瘤细胞类型数据。
从GTRD ChIP-Seq数据库中提取了所有收集到的细胞类型中的转录因子结合位点,这些位点要至少被三种peak calling算法检测到。如果转录因子结合到一个基因的近端调控区域(距离基因转录起始位点1kb以内的基因组区域),则认为该基因是转录因子的靶标。
2. 评估转录因子活性
使用上述5946个差异表达基因来计算转录因子活性。使用多元线性回归来评估每个样本中转录因子活性。保留超过5%的样本中活跃的转录因子进行进一步的分析。使用Limma分析癌症样本与正常前列腺组织转录因子活性的差异(FDR < 0.05)。
3. lncRNA转录调控的预测
使用上述5946个差异表达基因来预测lncRNA的转录调控。首先使用Pearson相关性分析计算每个基因和lncRNA表达值相关性。如果lncRNA满足以下两个标准,则认为该lncRNA以顺式方式调控基因:①表达相关性高(|coefficient| > 0.3,FDR < 0.01) ②其转录起始位点之间的最小距离小于1kb。如果Triplexator检测到lncRNA与靶标转录起始位点1 kb以内的DNA片段结合,在近端调节区形成一个RNADNA三联体,认为该lncRNA以反式反式调控靶基因。
然后预测转录因子与lncRNA之间的互作,限制差异表达基因是转录因子靶点,测量它们与lncRNA的表达相关性及其近端调控区转录因子结合位点数的Spearman相关性(FDR < 0.01)。
4. 生存分析和功能分析
根据表达值中值将样本分为高低组,使用R包survminer绘制Kaplan-Meier生存曲线,Log-rank 检验患者生存差异,使用R包survival的单变量Cox比例风险回归和R包glmnet的LASSO Cox回归评估风险比。使用Wald检验来计算单变量Cox回归模型的显著性(p < 0.05)。
使用R包clusterProfiler进行基因集富集分析(GSEA)和过表达分析
5. 统计分析
除了比较前列腺肿瘤和正常前列腺的转录因子活性以外,其余使用双尾t检验分析两组差异。使用单因素方差分析比较两组以上的差异,差异显著(p< 0.05),则采用Tukey’s HSD检验进行多重配对比较。
三、结果
1. 前列腺肿瘤和正常组织中转录因子的活性
转录因子在驱动和促进前列腺癌的起始和进展中的作用,它们的转录效应通常需要多种因素的合作,并依赖于细胞环境。该工作使用采用多变量回归模型来估计转录因子的活性,对每个样本进行多元线性回归分析,以转录因子占有率作为预测变量,以基因表达水平(zFPKM)作为结果变量(图1a)。为了确定回归模型能否推断出转录因子的活性,首先研究了两种研究充分的肿瘤蛋白MYC和TP53。将基因表达与MYC活性的相关性进行排列,发现特征基因出现在基因列表的顶部(图1b)。此外,MYC活性随基因拷贝数增加而增加(图1c)。而另一种转录因子TP53,其活性随着基因拷贝缺失而降低(图1c)。TP53突变导致其活性降低(图1d)。并非所有转录因子的活性都受到基因组拷贝数改变的影响。拷贝数变异可能改变基因表达水平,而表达水平并不是转录因子活性的唯一决定因素。这一事实可以解释基因拷贝数变异和转录因子活性之间的差异。
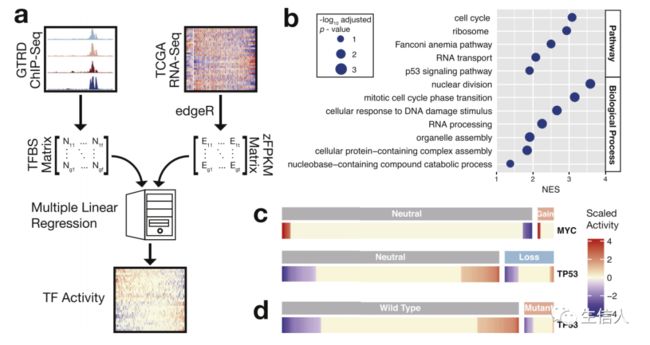
图1. 计算正常前列腺和原发性前列腺癌的转录因子活性
在前列腺癌中,发现67和63个转录因子的活性分别升高和降低(图2a-e)。原发肿瘤中TP53抑癌基因活性降低(图2b),MYC原癌基因活性升高(图2c)。肿瘤中发现AR活性增强(图2a),与之前的研究结果一致,即AR信号通路促进正常前列腺细胞和前列腺癌细胞生长和存活。AR在前列腺癌起始过程中作用的一个明显例子是雄激素依赖性的ERG激活,ERG过表达促进前列腺癌细胞浸润。该工作发现原发性肿瘤的ERG活性增加(图2d)。然后对上述在肿瘤和正常组织中差异活性的130个转录因子进行GO富集,几乎所有的富集条目与癌症有关(图2f),前列腺癌相关的富集在top。癌症中的转录失调通路上调(图2g)。
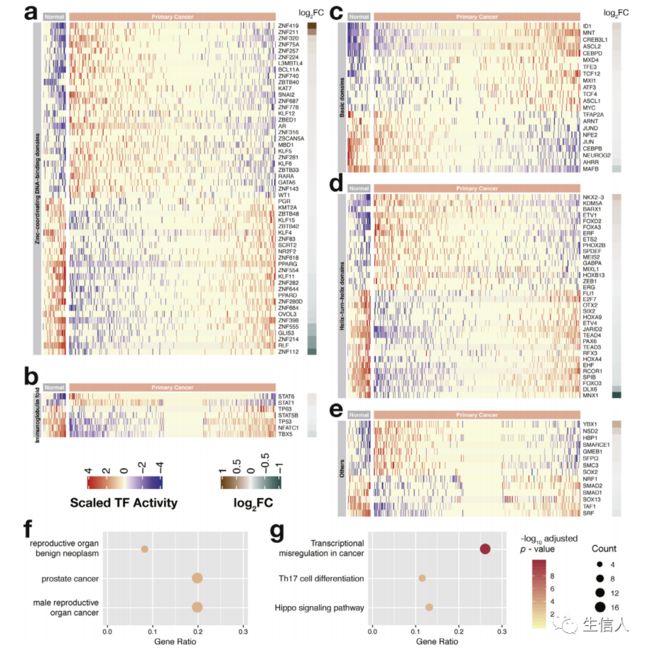
图2. 正常前列腺和原发性前列腺癌之间的转录因子活性差异
2. 转录因子与前列腺癌临床病理特征相关
接下来通过计算转录因子活性与前列腺癌临床病理因素之间的Pearson相关性,评估转录因子的临床价值。Gleason评分是前列腺癌的分级系统,分数越高越具有侵略性。共有26个转录因子和30个转录因子与组织学分级呈正相关和负相关(图3a-e)。16个转录因子和22个转录因子与T期呈正相关和负相关(图3f-j)。MYC等基因与组织学分级和临床分期正相关,AR等基因与两个临床病理参数呈负相关。发现晚期肿瘤中AR活性下降。最近的一项研究表明,过度活化的AR会诱导DNA双链断裂和细胞周期阻滞。
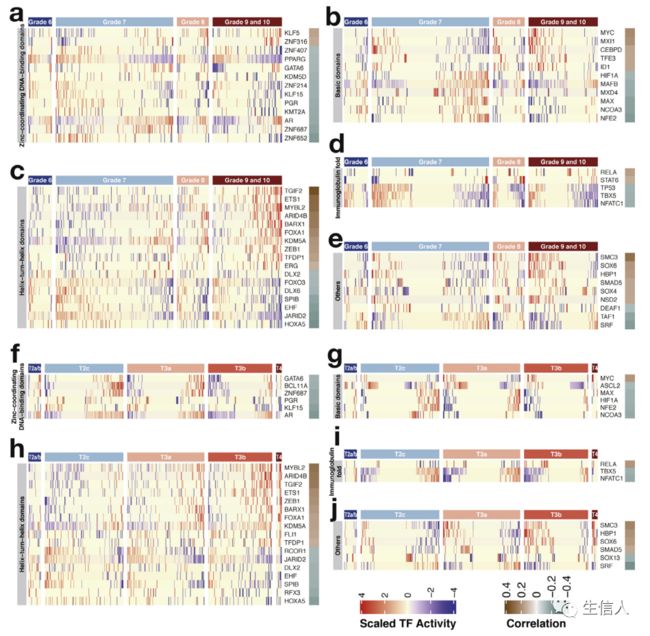
图3. 转录因子的活性与临床病理特征相关
3. 前列腺癌的预后转录因子
生存分析显示,ARID4B等20个转录因子的活性与前列腺癌复发正相关(图4a),JARID2等转录因子与好的预后相关。影响总生存率的潜在预后因素包括ARID4B等12个因子(图4b)。单变量Cox回归分析显示ARID4B等是不良预后因素,SRF等是好的预后因素(图4c)。接下来使用LASSO Cox回归模型去除对患者生存影响最小的转录因子,建立预后特征更好地预测临床结果。选择16个转录因子构建前列腺癌复发的预后标志物(图4d)。
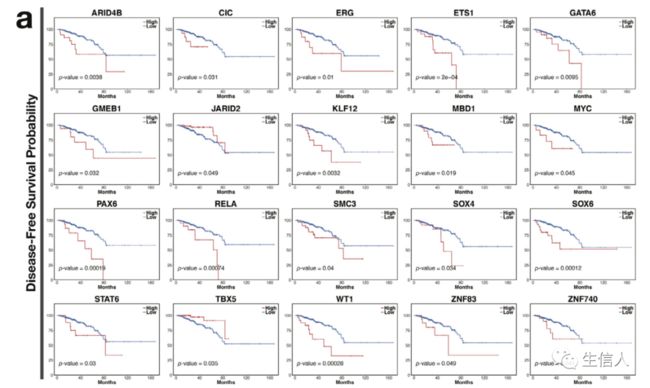
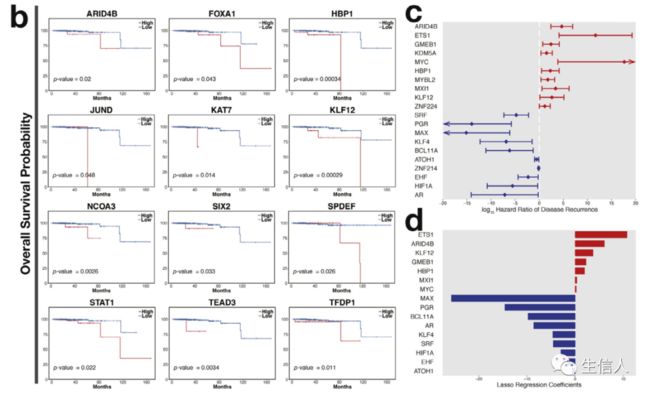
图4. 前列腺癌进展相关的潜在转录因子
根据16个基因的预后特征计算风险评分。研究发现前列腺癌风险评分与Gleason评分、临床分期、年龄、肿瘤细胞数量和PSA水平相关(图5a-e)。TCGA研究根据基因组数据定义了前列腺癌的7种亚型,但与风险评分无关。KM生存分析表明16个转录因子特征的风险评分能评估预后(图5f)。单因素和多因素Cox回归分析证实了16个转录因子特征的预后价值,并能独立预测患者的生存(图5g,h)。
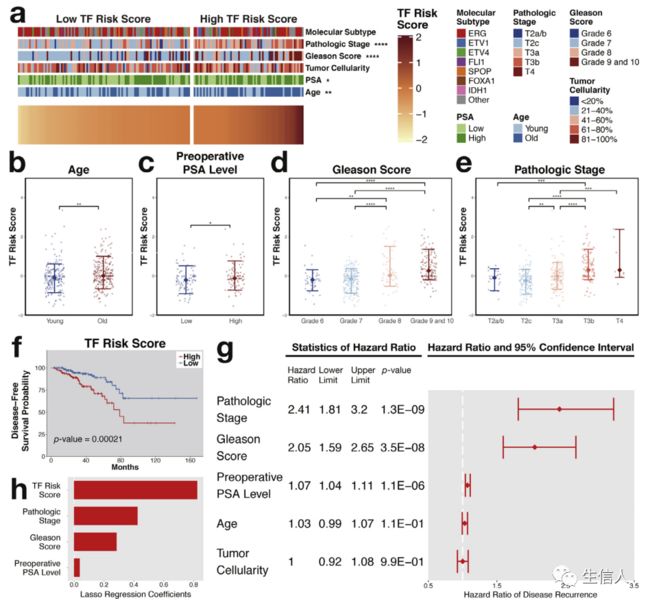
图5. 风险转录因子的预后价值
超过一半的预后标志转录因子与前列腺癌有关。MAX不仅与促癌因子相互作用,还与抑癌因子相互作用。除了癌症通路、G0和G1早期等通路以外(图6b),这些转录因子参与了腺体发育、上皮细胞形态发生等(图6a)。
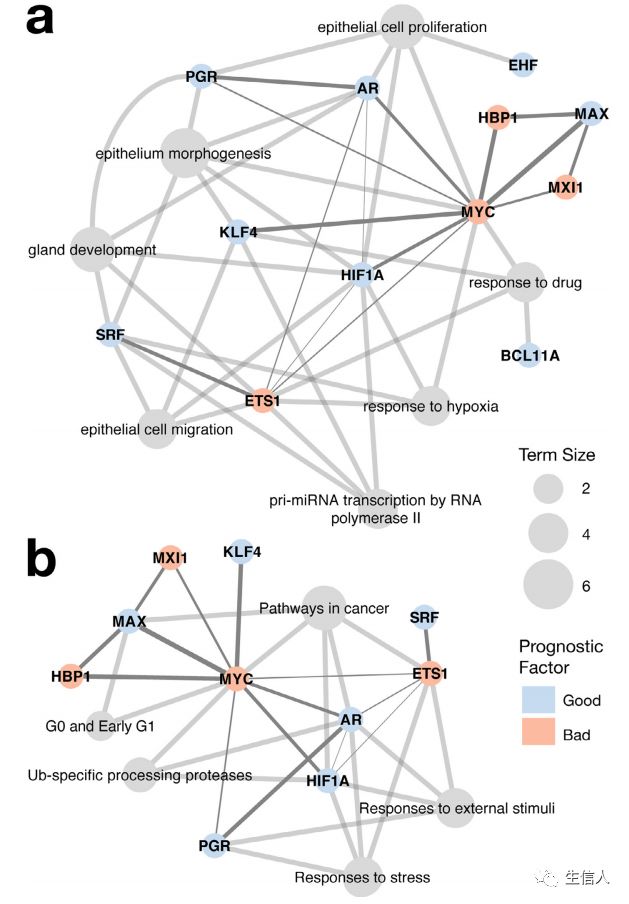
图6.预后特征的转录因子调控网络
4. 长非编码RNA在前列腺癌中的顺式和反式调控
另一种转录调节因子是长链非编码RNA,它可以作用于cis(其转录起始位点附近)或trans(其转录起始位点远端)。在包括前列腺癌在内的某些原发性癌症中,lncRNA作为转录调节因子参与了癌变。根据基因位点之间的表达相关性和距离,共识别了42个lncRNAs顺式调控,涉及41个lncRNAs。lncRNAs的反转录调控可能通过直接与基因组调控元件相互作用和/或与其他转录调控因子如转录因子结合来实现。
基于lncRNAs可以通过与转录因子相互作用来调控基因转录的假设,该工作开发了一个识别lncRNAs与转录因子相互作用的方法(图7a)。如果lncRNA和转录因子能够相互作用,那么它们应该具有共同的靶基因。在分析中,共同靶基因局限于转录因子靶基因,在前列腺癌中也表现出差异表达。获得了转录因子在共同靶基因近端调控区域的占据情况,以近似分析其对靶基因表达的影响。还计算了lncRNA表达水平与共同靶基因的Pearson相关性,以估计它们对靶基因的调控。最后,计算lncRNA调控与转录因子效应之间的Spearman’s rank相关性,分析它们之间的相互作用。共发现相互作用11398个,其中lncRNAs 839个,转录因子124个。
5. 特征转录因子-lncRNA互作支配转录调控
该工作还构建了一个数据库LNCTRN (https://navy.shinyapps.io/lnctrn),存储上述lncRNA参与的调控回路(图7b)。
接下来对预后转录因子及其lncRNA伙伴进行研究。在16个转录因子中,MYC和AR是显示高连接的hub基因(图6)。分别识别到201和147个lncRNA可能与MYC和AR相互作用。随机选择至少3个与MYC和AR相互作用的lncRNA进行实验验证(图7c-d)。三个MYC候选中的一个被证实与MYC发生物理相互作用(图7e)。AL590617.2表达随着肿瘤分级的升高而增加(图7c)。然后随机选取了4个AR的lncRNA伙伴(图7f),AL031714.1和AC110285.1在高级别肿瘤中表达升高,高AL031714.1水平的患者预后较差(图7d)。YBX1在LNCaP前列腺癌细胞中可与AR相互作用并作为AR激活剂发挥作用,共免疫共沉淀(图7g)证实AR与lncRNA HOXA10-AS、AC110285.1、TPM1-AS和AL031714.1相互作用,它们可能与AR协同调控AR靶点的转录。接下来进一步分析了MYC-AL590617.2的转录调控作用。对AL590617.2和MYC进行RNA干扰,检测与AL590617.2相关的前4个MYC靶基因的表达变化。AL590617.2和MYC敲低显著降低了两个靶点OXLD1和REX1BD的表达(图7h)。此外,MYC敲低降低了AL590617.2的表达水平。因此,MYC与AL590617.2可能形成前馈调节回路,是一种普遍存在的转录控制。
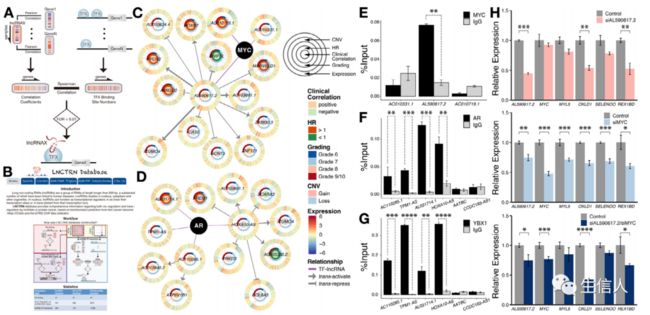
图7. 长非编码RNA与预后特征转录因子相互作用
总结:
该工作首先基于TCGA基因表达数据识别原发前列腺癌组织和正常组织的差异表达基因,然后使用多元线性回归来评估转录因子的活性,并识别在前列腺癌中活性显著上调和下调的转录因子。接下来评估了转录因子活性与临床病理学特征的相关性,并识别与预后相关的转录因子,构建COX回归模型并建立16个转录因子的预后模型。接下来开发了一个识别lncRNAs与转录因子相互作用的方法,识别特征转录因子-lncRNA互作并构建数据库LNCTRN。最后集中在几个具有代表性的预后转录因子及其lncRNA伙伴直接的关联进行实验分析。
意向分析(http://gaptechsxr.mikecrm.com/1vdMmqy)