scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
3-创建ArchRProject
An ArchRProject允许我们将多个Arrow文件组合到一个项目中。这ArchRProject很小,并存储在内存中。通过与进行交互,ArchRProject我们可以快速从Arrow文件中推送和提取数据。因此,它构成了几乎所有ArchR功能和分析工作流程的基础。此外,ArchRProject可以在以后保存和重新加载对象,从而提供了分析的连续性并促进了合作者之间共享分析项目。本部分介绍如何创建ArchRProject对象并与之交互。
3.1ArchRProject构建
首先,我们必须ArchRProject通过提供Arrow文件列表和一些其他参数来创建自己的文件。在outputDirectory这里描述了所有下游的分析和地块将被保存。ArchR将自动将先前提供的geneAnnotation和genomeAnnotation新的关联ArchRProject。这些是addArchRGenome("hg19")在上一章中运行时存储的。
projHeme1 <- ArchRProject(
ArrowFiles = ArrowFiles,
outputDirectory = "HemeTutorial",
copyArrows = TRUE #This is recommened so that if you modify the Arrow files you have an original copy for later usage.
)
使用addArchRGenome(Hg19)设置的GeneAnnotation!
使用addArchRGenome(Hg19)设置的GeneAnnotation!
正在验证箭头…
正在获取SampleNames…
将ArrowFiles复制到Ouptut目录!如果要节省磁盘空间,请设置copyArrows = FALSE
1 2 3
获取单元元数据…合并单元元数据…
初始化ArchRProject…
我们将其称为ArchRProject“ projHeme1”,因为它是我们造血工程的第一个迭代。在整个演练中,我们将对其进行修改和更新,ArchRProject并通过迭代项目编号(即“ projHeme2”)来跟踪正在使用的项目版本。
我们可以检查以下内容ArchRProject:
projHeme1
类:ArchRProject
输出目录: jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(11):示例TSSEnrichment…DoubletScore
DoubletEnrichment
numberOfCells(1):10661
平均TSS(1):16.832
平均碎片(1):3050
从上面我们可以看到我们ArchRProject已经初始化了一些重要的属性:
- 指定的
outputDirectory。 - 将
sampleNames其从该箭头的文件获得的每个样品的。 - 称为的矩阵
sampleColData,其中包含与每个样本相关的数据。 - 称为的矩阵
cellColData,其中包含与每个单元相关的数据。因为我们已经使用来计算doublet富集得分addDoubletScores(),该得分将这些值添加到Arrow文件中的每个单元格中,所以我们可以在cellColData矩阵中看到与“ DoubletEnrichment”和“ DoubletScore”相对应的列。 - 我们项目中的单元格总数,代表经过双重识别和去除后的所有样本。
- TSS富集度中值和所有细胞和所有样品中片段的中位数。
我们可以检查多少内存用于存储ArchRProjectR中的内存:
paste0("Memory Size = ", round(object.size(projHeme1) / 10^6, 3), " MB")
[1]“内存大小= 37.135 MB”
我们也可以询问在ArchRProject开始添加到该项目后哪些可用的数据矩阵对下游有用:
getAvailableMatrices(projHeme1)
[1]“ GeneScoreMatrix”“ TileMatrix”
3.2操纵ArchRProject
现在,我们已经创建了一个ArchRProject,我们可以做很多事情来轻松访问或操纵相关数据。
例子1. $访问器允许直接访问cellColData
我们可以访问与每个单元关联的单元名称:
head(projHeme1$cellNames)
[1] “scATAC_BMMC_R1#TTATGTCAGTGATTAG-1” “scATAC_BMMC_R1#AAGATAGTCACCGCGA-1”
[3] “scATAC_BMMC_R1#GCATTGAAGATTCCGT-1” “scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1”
[5] “scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1” “scATAC_BMMC_R1#AGTTACGAGAACGTCG-1”
我们可以访问与每个单元格关联的样本名称:
head(projHeme1$Sample)
[1]“ scATAC_BMMC_R1”“ scATAC_BMMC_R1”“ scATAC_BMMC_R1”“ scATAC_BMMC_R1”
[5]“ scATAC_BMMC_R1”“ scATAC_BMMC_R1”
我们可以访问每个单元的TSS丰富分数:
quantile(projHeme1$TSSEnrichment)
0%25%50%75%100%
4.027 13.922 16.832 19.937 41.782
例子2. ArchRProject用单元格子集
我们可以通过多种方法将an子集化,ArchRProject以仅获取一组选定的单元格。
我们可以按数字对项目进行子集化,例如,获取项目中的前100个单元格:
projHeme1[1:100, ]
类:ArchRProject
输出目录:jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(11):示例TSSEnrichment…DoubletScore
DoubletEnrichment
numberOfCells(1):100
平均TSS(1):10.7725
平均碎片(1):10200.5
我们可以根据某些单元格名称对项目进行子集化:
projHeme1[projHeme1$cellNames[1:100], ]
类:ArchRProject
输出目录: jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(11):示例TSSEnrichment…DoubletScore
DoubletEnrichment
numberOfCells(1):100
平均TSS(1):10.7725
平均碎片(1):10200.5
我们可以对项目进行子集化,以使所有单元格都与特定样本相对应:
idxSample <- BiocGenerics::which(projHeme1$Sample %in% "scATAC_BMMC_R1")
cellsSample <- projHeme1$cellNames[idxSample]
projHeme1[cellsSample, ]
类:ArchRProject
输出目录: jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(11):示例TSSEnrichment…DoubletScore
DoubletEnrichment
numberOfCells(1):4932
平均TSS(1):15.254
平均碎片(1):2771
我们可以将项目子集化,以仅保留满足TSS富集得分特定截止值的细胞:
idxPass <- which(projHeme1$TSSEnrichment >= 8)
cellsPass <- projHeme1$cellNames[idxPass]
projHeme1[cellsPass, ]
类:ArchRProject
输出目录: jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(11):示例TSSEnrichment…DoubletScore
DoubletEnrichment
numberOfCells(1):10500
平均TSS(1):16.9275
平均碎片(1):3042
例3.将数据添加到 ArchRProject
我们可以添加列以cellColData存储与项目相关的任何类型的特定于单元格的元数据。
例如,我们可以cellColData通过从原始样品名称中删除多余的信息,在其中添加一个包含更多清晰样品名称的列:
bioNames <- gsub("_R2|_R1|scATAC_","",projHeme1$Sample)
head(bioNames)
[1]“ BMMC”“ BMMC”“ BMMC”“ BMMC”“ BMMC”“ BMMC”
[7]“ BMMC”“ BMMC”“ BMMC”“ BMMC”“ BMMC”“ BMMC”
添加名为to的列的一种方法cellColData是直接使用$访问器。
projHeme1$bioNames <- bioNames
或者,我们可以cellColData使用addCellColData()函数添加一列。ArchR允许添加仅包含单元格子集信息的列。
bioNames <- bioNames[1:10]
cellNames <- projHeme1$cellNames[1:10]
projHeme1 <- addCellColData(ArchRProj = projHeme1, data = paste0(bioNames),
cells = cellNames, name = "bioNames2")
默认情况下,ArchR将使用填充缺少的条目NA。因此,当我们可以比较这两列时,我们看到NA在以下位置没有可用数据bioNames2:
getCellColData(projHeme1, select = c("bioNames", "bioNames2"))
具有10661行和2列的
DataFrame ## bioNames bioNames2
scATAC_BMMC_R1#TTATGTCAGTGATTAG-1 BMMC BMMC
scATAC_BMMC_R1#AAGATAGTCACCGCGA-1 BMMC BMMC
scATAC_BMMC_R1#GCATTGAAGATTCCGT-1 BMMC BMMC
scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1 BMMC BMMC
scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1 BMMC BMMC
......
scATAC_PBMC_R1#GCTGCGAAGATCCGAG-1 PBMC NA
scATAC_PBMC_R1#GCAGCTGGTGGCCTTG-1 PBMC NA
scATAC_PBMC_R1#GCAGATTGTACGCAAG-1 PBMC NA
scATAC_PBMC_R1#TTCGTTACATTGAACC-1 PBMC NA
scATAC_PBMC_R1#CGCTATCGTGAGGTCA-1 PBMC NA
例子4.从中获取列 cellColData
ArchR提供的getCellColData()功能使从中轻松恢复元数据列成为可能ArchRProject。
例如,我们可以按名称检索一列,例如每个细胞的唯一核(即非线粒体)片段数:
df <- getCellColData(projHeme1, select = "nFrags")
df
具有10661行和1列的
DataFrame
scATAC_BMMC_R1#TTATGTCAGTGATTAG-1 26189
scATAC_BMMC_R1#AAGATAGTCACCGCGA-1 20648
scATAC_BMMC_R1#GCATTGAAGATTCCGT-1 18991
scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1 18296
scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1 17458
......
scATAC_PBMC_R1#GCTGCGAAGATCCGAG- 1 1038
scATAC_PBMC_R1#GCAGCTGGTGGCCTTG-1 1037
scATAC_PBMC_R1#GCAGATTGTACGCAAG-1 1033
scATAC_PBMC_R1#TTCGTTACATTGAACC-1 1033
scATAC_PBMC_R1#CGCTATCGTGAGGTCA-1 1002
不用按名称选择列,我们实际上可以使用给定列的列名执行操作:
df <- getCellColData(projHeme1, select = c("log10(nFrags)", "nFrags - 1"))
df
具有10661行和2列的
DataFrame ## log10(nFrags)nFrags-1
scATAC_BMMC_R1#TTATGTCAGTGATTAG-1 4.4181189156542 26188
scATAC_BMMC_R1#AAGATAGTCACCGCGA-1 4.31487799153581 20647
scATAC_BMMC_R1#GCATTGAAGATTCCGT-1 4.27854783377585 18990
scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1 4.26235615159869 18295
scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1 4.24199448915678 17457
......
scATAC_PBMC_R1#GCTGCGAAGATCCGAG-1 3.01619735351244 1037
scATAC_PBMC_R1#GCAGCTGGTGGCCTTG-1 3.01577875638904 1036
scATAC_PBMC_R1#GCAGATTGTACGCAAG-1 3.01410032151962 1032
scATAC_PBMC_R-1
GC#CA1CGCG-1AT#CG1CA1CG#AC1CTC#CG1CTC#1CA1CA1C#CG1C#AC1C#CFA#1
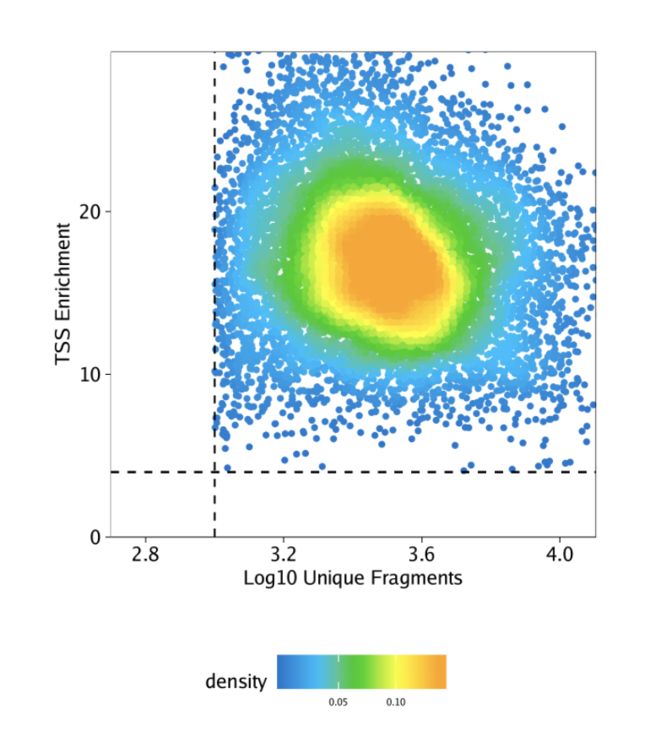
例子5.绘制QC指标-log10(唯一片段)与TSS富集得分
重复上面显示的示例,我们可以轻松获取用于单个细胞质量控制的标准scATAC-seq指标。我们已经发现,最有效的质量控制指标是TSS富集得分(ATAC-seq数据中信噪比的量度)和唯一核片段的数量(因为片段很少的细胞没有足够的数据自信地进行分析)。
df <- getCellColData(projHeme1, select = c("log10(nFrags)", "TSSEnrichment"))
df
具有10661行和2列的
DataFrame ## log10(nFrags)TSSEnrichment
scATAC_BMMC_R1#TTATGTCAGTGATTAG-1 4.4181189156542 7.149
scATAC_BMMC_R1#AAGATAGTCACCGCGA-1 4.31487799153581 7.911
scATAC_BMMC_R1#GCATTGAAGATTCCGT-1 4.27854783377585 4.505
scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1 4.26235615159869 6.946
scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1 4.24199448915678 4.799
......
scATAC_PBMC_R1#GCTGCGAAGATCCGAG-1 3.01619735351244 24.356
scATAC_PBMC_R1#GCAGCTGGTGGCCTTG-1 3.01577875638904 22.537
scATAC_PBMC_R1#GCAGATTGTACGCAAG-1 3.01410032151962 20.146
scATAC_PBMC_R1#TTCGTTACATTGAACC-1 3.01410032151962 30.198
scATAC_PBMC_R1#CGCTATCGTGAGGTCA-1 3.00086772153123 21.485
现在让我们根据TSS富集得分绘制唯一核片段的数量(log10)。这种类型的绘图是识别高质量单元格的关键。您会注意到,我们先前在创建Arrow文件(通过filterTSS和filterFrags)时指定的边界已经去除了低质量的单元格。但是,如果我们注意到先前应用的QC过滤器不足以满足该样本的需要,则可以根据该图进一步调整截止值,或者在需要时重新生成Arrow文件。
p <- ggPoint(
x = df[,1],
y = df[,2],
colorDensity = TRUE,
continuousSet = "sambaNight",
xlabel = "Log10 Unique Fragments",
ylabel = "TSS Enrichment",
xlim = c(log10(500), quantile(df[,1], probs = 0.99)),
ylim = c(0, quantile(df[,2], probs = 0.99))
) + geom_hline(yintercept = 4, lty = "dashed") + geom_vline(xintercept = 3, lty = "dashed")
p
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(p, name = "TSS-vs-Frags.pdf", ArchRProj = projHeme1, addDOC = FALSE)
[1]“绘制ggplot!”
[1] 0
3.3绘制来自ArchRProject的样本统计信息
在单个集成数据集中处理多个不同样本时,比较所有样本中的各种指标可能很重要。ArchR为分组数据提供了两种主要的绘图机制:山脊绘图和小提琴绘图。这些都可以通过plotGroups()函数访问。当然,这种绘图类型不仅限于样本级别的数据,还可以用于绘制诸如群集之类的组的下游组级别信息。
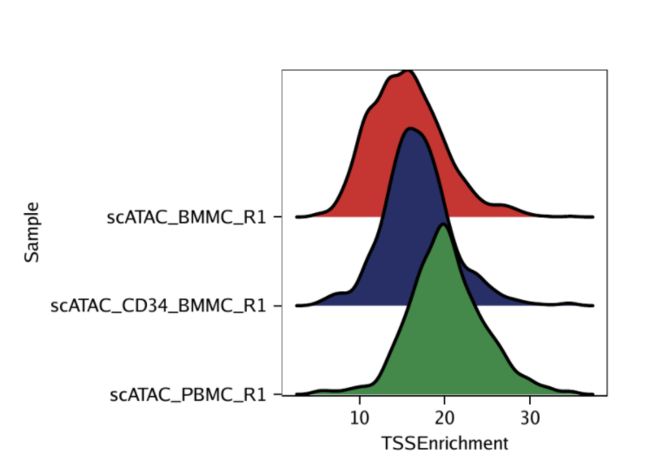
示例1.为TSS富集得分的每个样品绘制一个山脊图。
要绘制山脊图,我们设置plotAs = "ridges"。
p1 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "TSSEnrichment",
plotAs = "ridges"
)
p1
拾取联合带宽为0.882
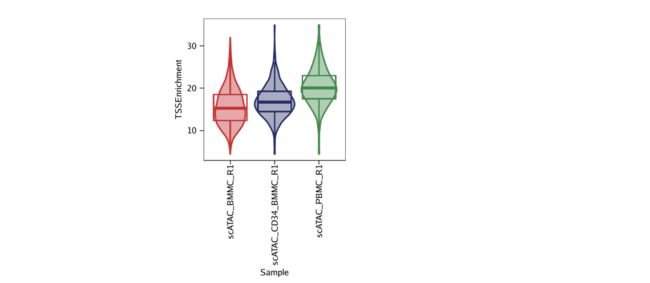
示例2.为每个样品绘制一个小提琴图,以获取TSS富集得分。
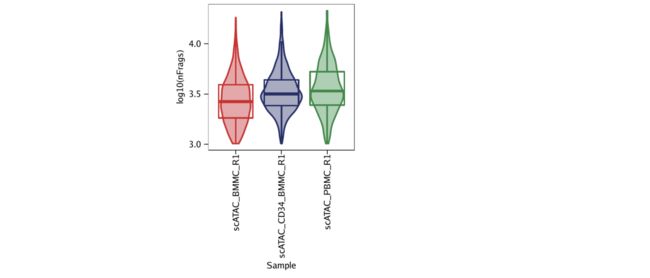
要绘制小提琴图,我们设置plotAs = "violin"。ArchR中的小提琴图带有由实施的Tukey风格的盒须图ggplot2。这意味着下部和上部铰链分别对应于第25个和第75个百分位数,而中间铰链则对应于中间值。上下晶须从铰链延伸到最低或最高值,或者是四分位间距(第25个百分位数与第75个百分位数之间的距离)的1.5倍。
p2 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "TSSEnrichment",
plotAs = "violin",
alpha = 0.4,
addBoxPlot = TRUE
)
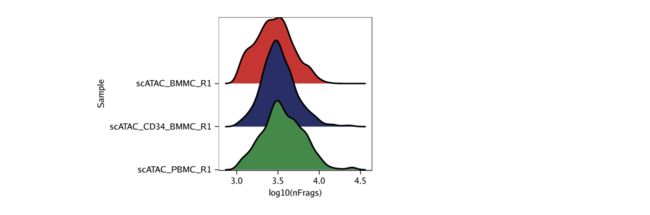
示例3.为log10(唯一核碎片)的每个样本绘制一个岭图。
p3 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "log10(nFrags)",
plotAs = "ridges"
)
示例4.为log10(唯一核碎片)的每个样本绘制小提琴图。
p4 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "log10(nFrags)",
plotAs = "violin",
alpha = 0.4,
addBoxPlot = TRUE
)
要保存这些图的可编辑矢量化版本,我们使用
plotPDF()。
plotPDF(p1,p2,p3,p4, name = "QC-Sample-Statistics.pdf", ArchRProj = projHeme1, addDOC = FALSE, width = 4, height = 4)
3.4绘制样品片段大小分布和TSS富集图。
由于数据的存储和访问方式,ArchR可以非常快速地从Arrow文件中计算片段大小分布和TSS富集配置文件。
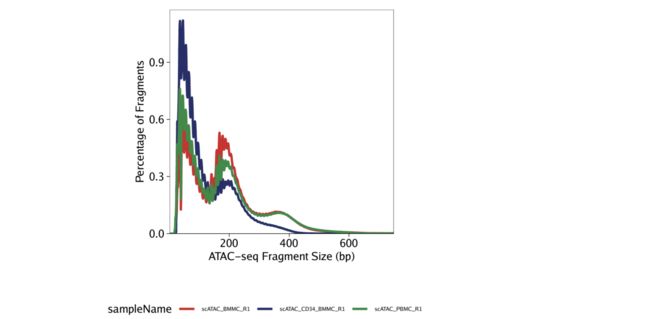
片段大小分布 要绘制所有样本的片段大小分布,我们使用plotFragmentSizes()函数。ATAC-seq中的片段大小分布在样品,细胞类型和批次之间可能存在很大差异。如下所示的轻微差异是常见的,不一定与数据质量的差异相关。
p1 <- plotFragmentSizes(ArchRProj = projHeme1)
p1
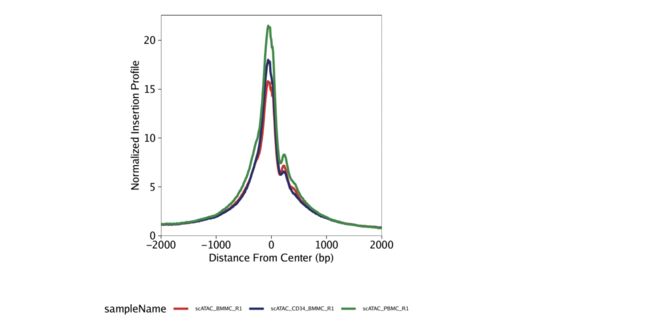
TSS富集曲线 要绘制TSS富集曲线,我们使用该plotTSSEnrichment()功能。TSS富集图谱应在中心显示一个清晰的峰,在中心右侧显示一个较小的肩峰,这是由位置良好的+1核小体引起的。
p2 <- plotTSSEnrichment(ArchRProj = projHeme1)
p2
要保存这些图的可编辑矢量化版本,我们使用
plotPDF()。
plotPDF(p1,p2, name = "QC-Sample-FragSizes-TSSProfile.pdf", ArchRProj = projHeme1, addDOC = FALSE, width = 5, height = 5)
3.5保存和加载ArchRProject
ArchR提供了一种简便的方法来保存ArchRProject对象,以便以后重新加载或与其他用户共享。从根本上讲,一个ArchRProject箭头指向一组Arrow文件。因此,ArchRProject使用saveArchRProject()函数保存的过程将:
- 将当前的Arrow文件复制到指定的文件,
outputDirectory以便它们与新ArchRProject对象专门关联。 - 将指定的副本保存
ArchRProject在中outputDirectory。
例如,我们可以保存projHeme1使用情况saveArchRProject(),这将使我们可以在以后的章节中使用该项目。
saveArchRProject(ArchRProj = projHeme1, outputDirectory = "Save-ProjHeme1", load = FALSE)
Copying ArchRProject to new outputDirectory : /oak/stanford/groups/howchang/users/jgranja/ArchRTutorial/ArchRBook/BookOutput4/Save-ProjHeme1
Copying Arrow Files…
Copying Arrow Files (1 of 3)
Copying Arrow Files (2 of 3)
Copying Arrow Files (3 of 3)
Getting ImputeWeights
No imputeWeights found, returning NULL
Copying Other Files…
Copying Other Files (1 of 1): Plots
Saving ArchRProject…
这将复制Arrow文件并将projHeme1 ArchRProject对象的.RDS文件保存在指定的中outputDirectory。很重要!此过程不会自动更新ArchRProject当前R会话中处于活动状态的对象。具体来说,projHeme1在当前R会话中命名的对象仍将指向Arrow文件的原始位置,而不是位于指定位置中的复制的Arrow文件outputDirectory。如果要执行此操作,我们将指定load = TRUE导致saveArchRProject()函数返回保存的ArchRProject对象的对象,您可以分配该ArchRProject对象以使用覆盖原始对象<-。这样可以有效地保存和ArchRProject从其新位置加载。
3.6从ArchRProject过滤双胞比率
在使用添加了预测双胞的信息之后addDoubletScores(),可以使用删除这些预测双胞filterDoublets()。此过滤步骤的关键要素之一是,filterRatio这是基于通过过滤器单元的数量,预测的双胞与过滤器的最大比率。例如,如果有5000个单元,则过滤后的预测双峰的最大数量为filterRatio * 5000^2 / (100000)(简化为filterRatio * 5000 * 0.05)。这filterRatio使您可以对可能具有不同双峰百分比的多个不同样品应用一致的过滤器,因为它们是在不同的细胞上样浓度下运行的。越高filterRatio,可能被去除为双胞的细胞数量越多。
首先,我们过滤双胞。ArchRProject在本逐步教程中,我们将其另存为新文件,但是您始终可以覆盖原始ArchRProject对象。
projHeme2 <- filterDoublets(projHeme1)
从ArchRProject过滤410个单元!
scATAC_BMMC_R1:4932的243(4.9%)
scATAC_CD34_BMMC_R1:3275的107(3.3%)
scATAC_PBMC_R1:2454的60(2.4%)
以前,我们看到projHeme1有10,661个细胞。现在,我们看到projHeme2有10,251个细胞,表明如上所述通过双峰过滤除去了410个细胞(3.85%)。
projHeme2
类:ArchRProject
输出目录: jgranja / ArchRTutorial / ArchRBook / BookOutput4 / HemeTutorial
样品(3):scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
sampleColData名称(1):ArrowFiles
cellColData名称(13):示例TSSEnrichment…bioNames bioNames2
numberOfCells(1):10251
平均TSS(1):16.856
平均碎片(1):2991
如果要从ArchR项目中过滤更多单元,则可以使用更高的filterRatio。要查看可以调整的其他参数,请尝试?filterDoublets。
projHemeTmp <- filterDoublets(projHeme1, filterRatio = 1.5)
从ArchRProject过滤614个单元!
scATAC_BMMC_R1:4932中的364(7.4%)
scATAC_CD34_BMMC_R1:3275中的160(4.9%)
scATAC_PBMC_R1:2454中的90(3.7%)
由于projHemeTmp仅出于说明目的而创建,因此我们将其从R会话中删除。
rm(projHemeTmp)
参考材料:
https://www.archrproject.com/