Title:Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment
单细胞测序揭示复杂的乳腺癌免疫微环境
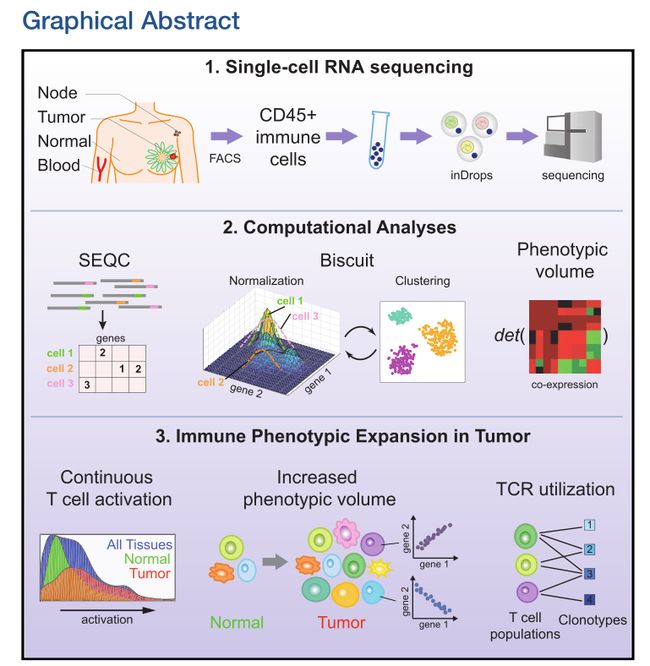
全文摘要:了解肿瘤微环境中免疫细胞表型对于深入理解肿瘤进展机制和免疫治疗反应至关重要。在该研究中,研究者采集8个未经治疗的乳腺癌患者(包括ER+、PR+、HER2+和TNBC患者)的临床样本(包括肿瘤组织、正常乳腺组织,外周血和淋巴结),利用荧光激活细胞分选(FACS)纯化的CD45 +细胞,使用inDrop平台对其进行单细胞RNA测序,总共构建了47016个CD45+细胞(CD45分子在所有白细胞上都有表达,称为白细胞共同抗原)的RNA测序数据集。接着进行计算机分析,使用SQEC pipeline,得到最终分析的基因表达矩阵;开发biscuit算法,在对输入数据进行归一化的同时进行聚类分析,然后将得到的聚类进行注释,得到多个免疫细胞亚基;定义“表型体积”,用于探究是否肿瘤组织内存在免疫表型扩增。结论部分,作者发现T细胞激活状态存在连续型;相比于正常组织,肿瘤组织内的免疫表型体积增加;利用额外27000个T细胞的配对RNA测序和TCR测序,揭示TCR的利用对免疫表型多样性的影响。
文章概览:本篇文章结果部分总共分为10个部分。前1-5部分主要从不同层面,展示单个患者单细胞测序结果、所有肿瘤样本汇总结果、所有样本汇总结果的情况,用于说明biscuit算法的可行性以及组织环境对免疫多样性的影响。由于聚类结果中主要是T细胞和骨髓源性细胞,故接下来第6-9部分和第10部分分别展示T细胞和骨髓源性细胞的详细情况。
结果部分:
Part 01:乳腺癌组织定居性免疫细胞的单细胞RNA测序
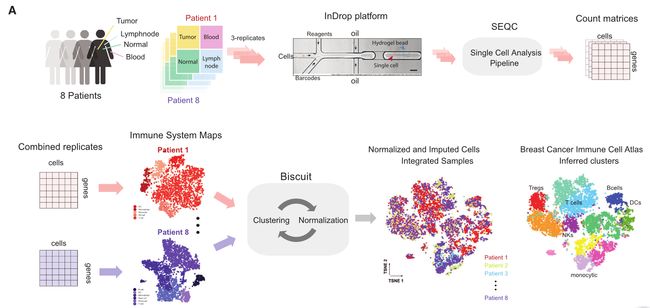
Fig 1A:工作流程:样本采集→单细胞平台测序→构建测序数据集→计算机算法处理与分析→结果可视化
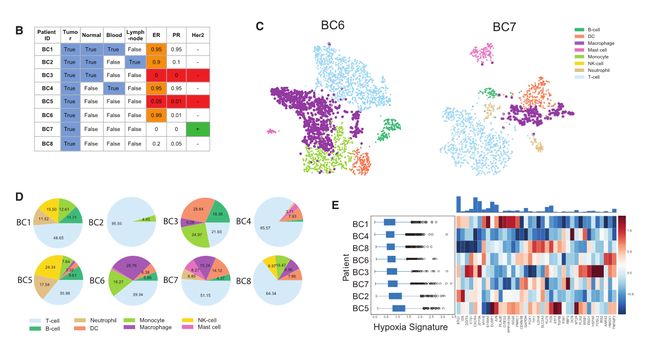
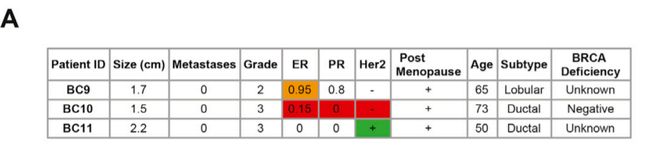
Fig 1B: 8个未经治疗的乳腺癌患者(包括ER+,PR+,HER2+和TNB患者)的临床样本信息。
Fig 1C: 进行每例乳腺癌患者测序结果的t-SNE投射。附图展示其余6位患者的t-SNE结果。每个点代表由表型聚类着色的单个细胞,并且用推断的细胞类型标记聚类。通过患者间的结果比较,可以看出患者间免疫细胞的组成和占比是由很大差异的。
Fig1D:与最近的研究一致,每位患者肿瘤组织中的免疫组成存在很大程度的变异。例如,骨髓和T细胞部分构成4 %-55%和21%-96%。
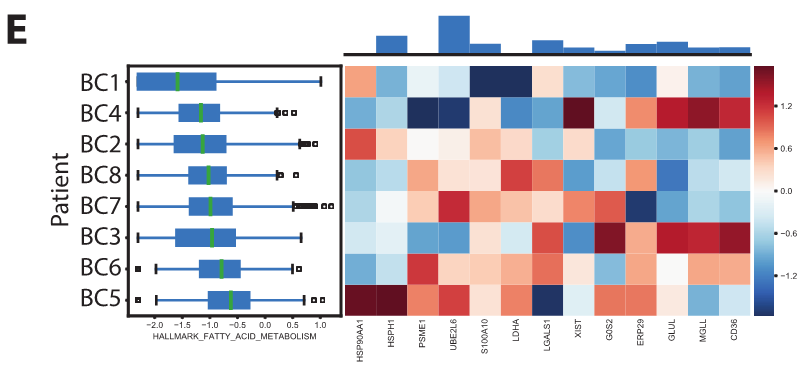
Fig1E:展示不同通路的基因表达情况,示患者间代谢特征的变异,包括缺氧(图1E)、脂肪酸代谢(SF1E)、糖酵解(SF1F)和磷酸化(SF1G)。
方框图(左)显示每个患者免疫细胞中每个特征的表达(定义为每个特征中基因的平均标准化表达);热图(右)显示每个特征中基因的Z评分平均表达;(上)条形图显示所有患者热图中显示的每个基因的表达。尽管所有患者在缺氧特征中表达相似的平均基因程度,但在特征中单个基因的水平表达不同。
Part 02: 开发biscuit算法,用于全部肿瘤样本数据的整合
为了能够系统比较患者间免疫细胞组成的差异,研究者合并所有患者的肿瘤样本测序数据。

Fig 2A:批次效应探究——基于文库大小标准化后,同一患者的细胞更倾向聚集在一起(左图)。基于Biscuit算法标准化后,能够使患者间的细胞混合程度提高,校正非细胞因素和批次效应。
分层贝叶斯模型Biscuit算法,其可在对输入数据归一化的同时推断聚类,能够校正细胞和批次内在变异。(右图)在Biscuit标准化和归类后得到8个乳腺肿瘤的免疫细胞的t-SNE图谱,显示患者间的细胞数据混合程度更高,显示出丰富的聚类。
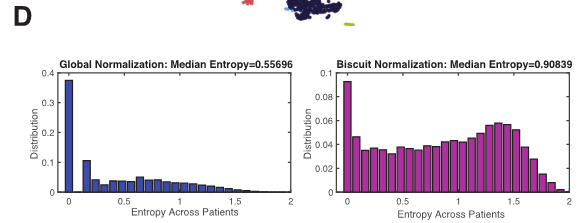
Fig 2D: 补充fig 2A, 基于熵的测量来量化细胞的混合程度(熵越大,混合程度越高):左边是文库大小标准化的结果,熵值较小(中位熵值0.55696),说明单个样本中细胞最相似,右图展示的是 Biscuit标准化后的结果,熵值增大(中位熵值0.90839),biscuit显着改善了患者间细胞混合程度(U = 1.7721 x 10- 9,p = 0)。
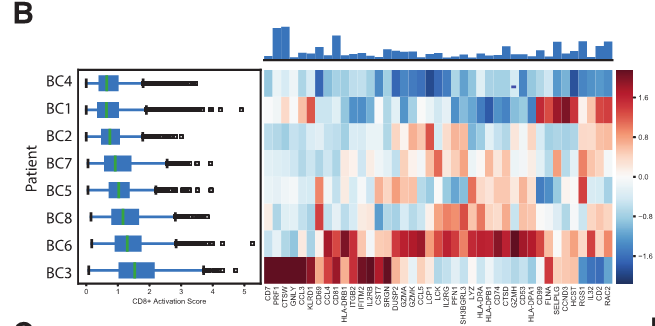
Fig 2B:每个患者的CD8T细胞活化特征:存在CD8 T细胞活化梯度,在TNBC肿瘤(BC3)中最明显。T细胞活化特征的梯度表达显示患者间的可变性。
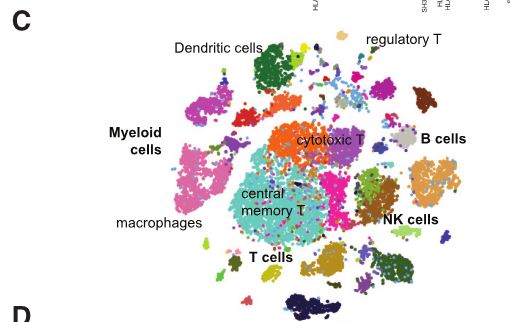
Fig 2C: 使用biscuit方法,成功保留了免疫细胞活化的信息,同时稳定了文库大小的差异,在输入数据中发现了丰富的结构,注释后发现T细胞,巨噬细胞,单核细胞,B细胞和自然杀伤(NK)细胞簇,表明瘤内免疫细胞亚型的多样性。
PART 03 :乳腺肿瘤免疫细胞图谱显示细胞状态的多样性
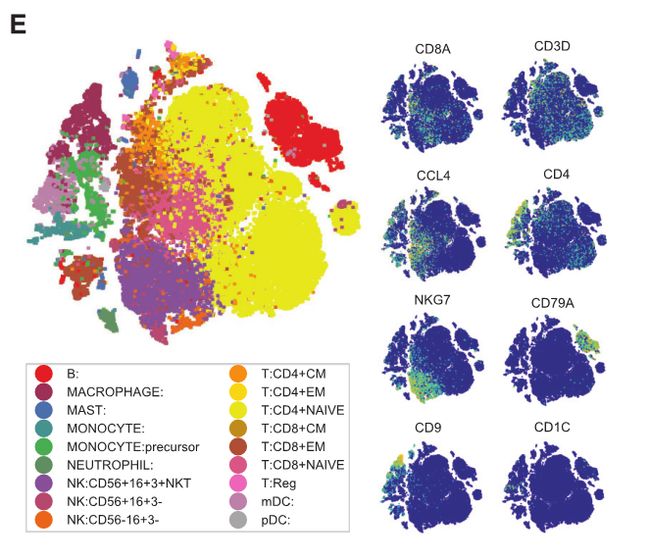
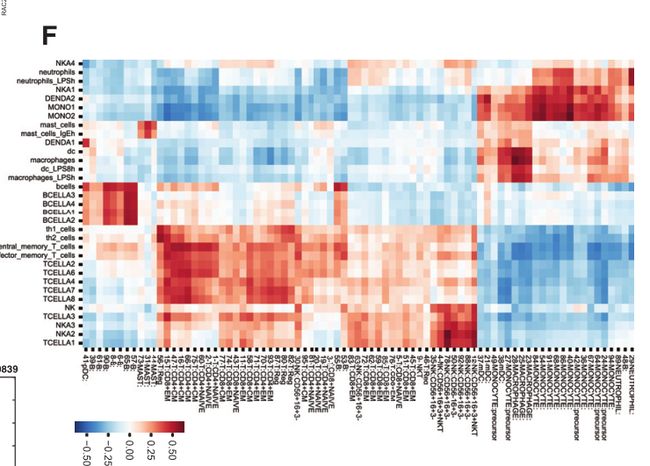
Fig 2E: 为了构建整体免疫细胞图谱,合并肿瘤,血液,淋巴和对侧正常组织的免疫细胞的全部测序数据,biscuit算法处理后的完整图谱t-SNE,通过推断细胞类型标志揭示了83个簇。
Fig 2F:对每一个簇进行注释,确定了38个T细胞、27个髓系细胞、9个B细胞和9个NK细胞簇。方法:将簇平均表达水平(横坐标)与纯化免疫细胞的批量RNAseq数据集进行了pearson相关分析。
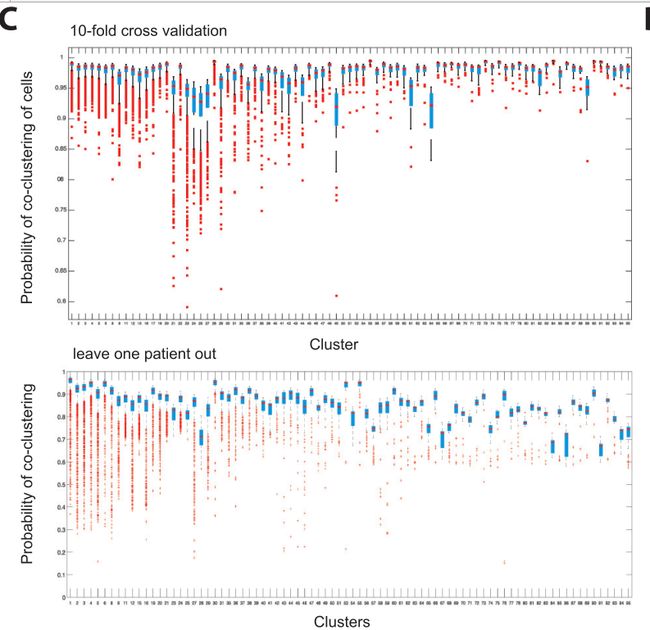
SF 2C:交叉验证测试聚类的稳健性,一是十折交叉验证,二是留一法,结果支持前面的聚类结果(随机选取10个总体数据的亚集重复上述分析,并进行聚类;去掉一个患者所用样本数据后,重复上述分析,并聚类),显示了稳健的集群分配能力。
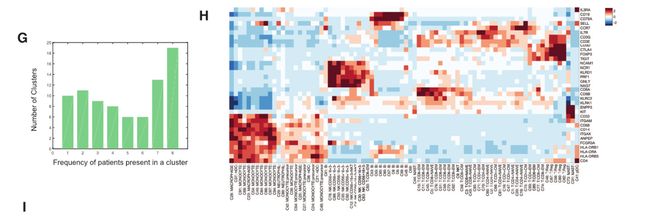
Fig 2G:大多数聚类在多个患者中共享,只有10个是患者特异性的。
Fig 2H: 对各簇中传统的细胞类型标志表达进行Z-评分标准化表达,确认这些簇与细胞类型之间的关系。在T细胞簇中,鉴定出15个CD8+和21个CD4+簇,被细分成9个幼稚、7个中枢记忆、15个效应记忆和5个Treg簇。髓样细胞簇分为3个巨噬细胞、3个肥大细胞、4个中性粒细胞、3个树突状细胞、1个浆细胞样树突状细胞和13个单核细胞簇。
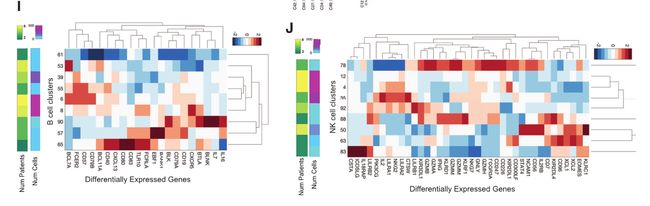
Fig 2I-2J: B细胞(左)和NK细胞(右)簇中的差异表达基因,由细胞类型中的Z 值标准化,以突出标记物与平均B或NK簇相比具有更高或更低表达的簇。最后确定了3个CD56-NK细胞簇和6个CD56+NK细胞簇,其中2个可能是NKT细胞。
由于T细胞和髓细胞代表了最丰富的细胞亚群,被认为是最具临床影响的细胞,故接下来集中对这些主要细胞类型进行了深入分析。
PART 04 : 组织环境对免疫表型多样性的影响
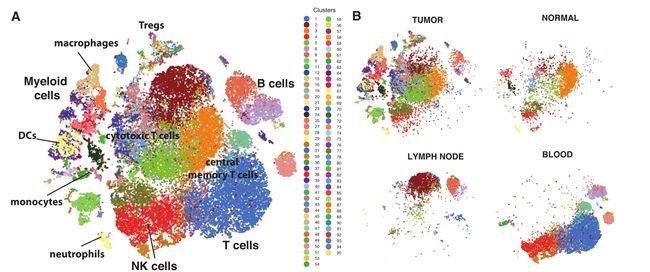
Fig 3A-3B: 使用t-分布随机邻域嵌入(t-SNE)来可视化组织间的表型重叠,量化免疫表型的变化在多大程度上是由组织环境驱动的。结果表明乳腺组织中的T细胞与血液中的t细胞、淋巴结中的t细胞表现出不同的表型。虽然正常组织和肿瘤样本之间显示出相当大的重叠,但我们观察到肿瘤中的表型异质性和细胞群的扩大。
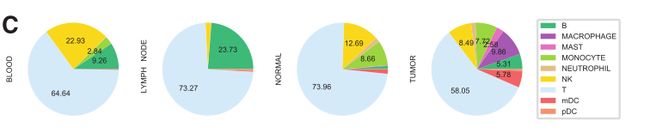
Fig 3C:各种组织中免疫细胞的组成比例。
原始T细胞在3个血液特异性簇(,P=3x 10-80);
b细胞在淋巴结中比在其他组织中更为普遍(,p=0.0);
肿瘤组织和正常组织中均存在T细胞簇的亚群,但肿瘤中细胞毒性T细胞簇更为丰富(,p=3 x 10-25),Treg簇也一样(,p=5x10-91)。
正常组织和肿瘤组织之间共享一些髓样细胞簇,而更活跃的巨噬细胞簇(TAMs)对肿瘤具有特异性(,p=0.0)。
这些发现强调了组织环境是免疫表型的重要决定因素,基于血液免疫细胞的生物标志物可能不一定反映肿瘤中的免疫细胞组成。
PART 05: 免疫细胞在肿瘤微环境中进行表型扩增
大量正常乳腺组织中存在免疫细胞状态,包括在循环或次级淋巴组织脾、胸腺等中未观察到的13个髓样细胞和19个T细胞簇。在正常乳腺组织存在的免疫细胞种类,同样也是肿瘤组织免疫细胞种类中的一部分,但肿瘤组织中有独特的免疫细胞簇,包括14个髓样细胞簇和17个T细胞簇。相比之下,正常组织没有特异性簇。前面的结果,提示肿瘤组织相对于正常组织的细胞聚类群体更多、异型性更大。
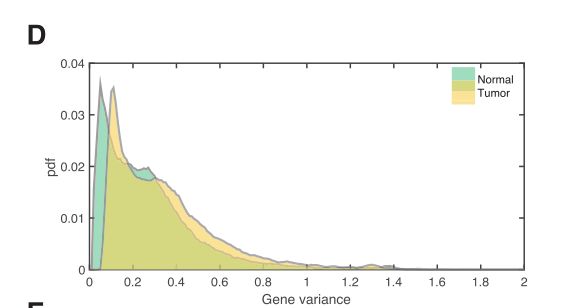
Fig 3D: 对正常乳腺组织和肿瘤组织的所有免疫细胞(所有患者)的每个基因计算表达差异的分布。与正常组织相比,肿瘤中基因表达的差异显著增加,从而导致细胞状态的多样性增加。
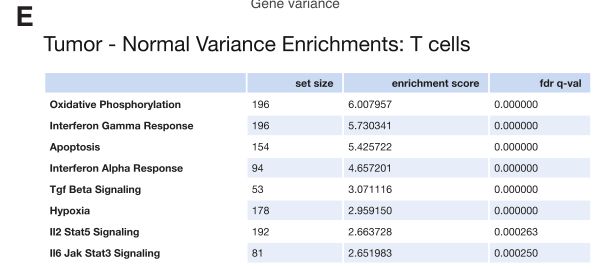
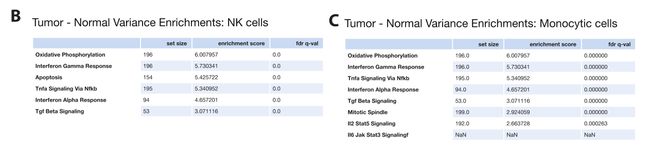
分别对T细胞(Fig 3E)、NK(SF 3B)和单核细胞(SF 3C)中显著差异表达的基因进行基因集富集分析(GSEA),主要在一下通路中富集: 包括I型干扰素(IFNα)和II型干扰素(IFNγ)、肿瘤坏死因子α(TNF-α)、转化生长因子β(TGF-β)、IL6/JAK/STAT信号、缺氧等通路。
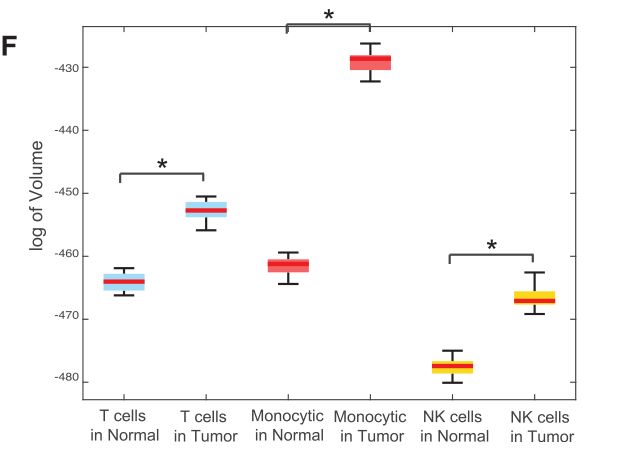
Fig 3F: 与正常乳腺组织相比,肿瘤中主要细胞类型(包括T细胞、髓细胞和NK细胞)的表型体积显著增加。
与正常组织相比,肿瘤组织中的基因表达变异增加。是什么原因导致肿瘤组织中表型多样性的原因呢?研究者猜想,是否肿瘤组织中有额外的活动过程或者表型的激活,使其异于正常组织。为了进一步探讨这种差异的增加,研究者定义了一种细胞“表型体积”的度量:用基因表达协方差来度量独立表型的容积扩增。
具体计算:如果肿瘤组织有更多的与正常组织不同的激活表型,即独立的表型,那么其表型体积越大。举个例子,如果gene i 与其他gene 的协方差模式与gene I’与其他基因的协方差模式相似,那么gene i 和gene I’就是非独立的,gene i不增加表型体积,反之则增加表型体积。用此方法处理所有的基因,用于评估肿瘤组织中激活的表型的表达变异是否独立于正常组织。如果在肿瘤组织中发现新的独立表型,可以提示肿瘤组织中存在激活的其他机制和通路。
利用这个指标,研究者比较了正常组织和肿瘤组织中每种细胞类型所占的表型体积。T细胞、髓细胞和NK细胞的体积倍数变化分别为7.39x104、1.18x1014和6.08x104,表明肿瘤的表型体积比正常组织大幅度增加。这些数据表明,肿瘤内细胞状态的异质性增加和显著的表型扩展可能是由于肿瘤内的局部微环境更加多样,其炎症、缺氧程度、激活和抑制受体配体的表达以及营养供应不同。
PART 06: 瘤内T细胞具有变异的连续成分
接着使用扩展映射来描述表型变异的重要来源。
扩展映射:通过整合数据的局部几何关系,解释数据集在不同尺度的几何结构;与PCA、MDA这些降维方法相比,扩展映射为非线性,聚焦于发现数据集潜在的流形结构
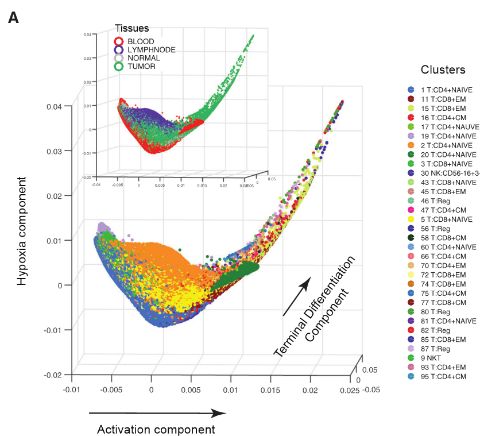
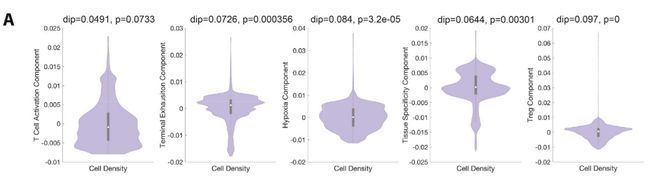
Fig 4A;SF4A: 大多数成分T细胞成分定义了逐渐变化的趋势。前三大信息成分分别与激活、终末分化和缺氧信号相关。
左图:每个点代表一个细胞,该细胞按簇类型着色。 主要轨迹用箭头指示,并用与每个组件最相关的标志进行注释。
右图:小提琴图展示沿各组分轴的细胞分布情况。
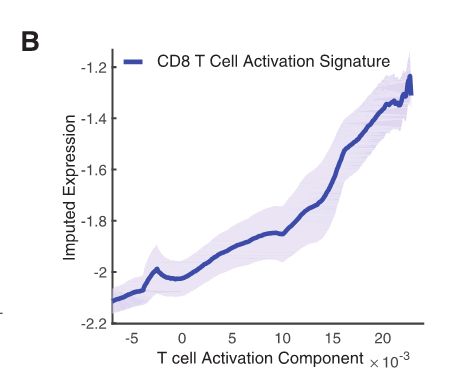
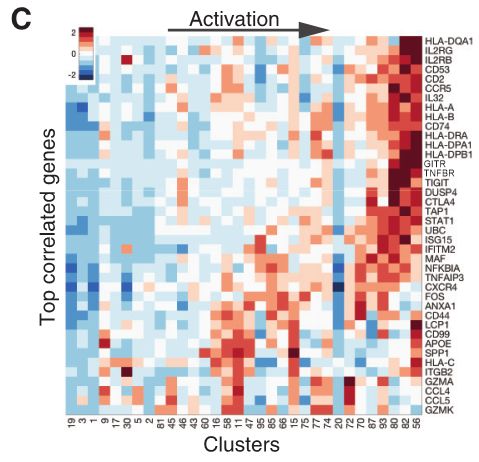
Fig 4B-4C: 第一大信息组分-“T细胞激活组分”。
标记为“激活”的信息量最大的成分与T细胞激活和进行性分化的基因特征以及IFNγ信号(p=0.0)高度相关。激活信号的平均表达量沿着T细胞激活组分逐渐增加(左图),同时特定激活相关基因的表达逐渐增加(右图)。
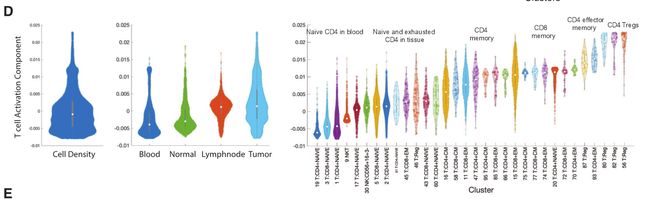
Fig 4D:小提琴图显示了所有T细胞(左),单个组织(中)和单个簇(右)中沿着激活成分的T细胞的密度。 每个小提琴内的点数与单元数成正比。
肿瘤内T细胞群,包括Treg和效应记忆T细胞,在组分激活端富集(t检验,p=0.0),而原始外周血T细胞聚集在激活最弱的末端,与静息状态一致(t检验,p=0.0)。尽管簇的平均表达水平随着组分的变化而逐渐变化,但在每个簇中激活状态变化范围较大(右)。已知与该成分最相关的基因随激活和进行性分化增加,包括细胞溶解效应分子颗粒酶A和K(GZMA和GZMK)、促炎细胞因子(IL-32)、细胞因子受体亚单位(IL2RB)、趋化因子(CCL4、CCL5)和趋化因子受体(CXCR4、CCR5)(fig 4C)。
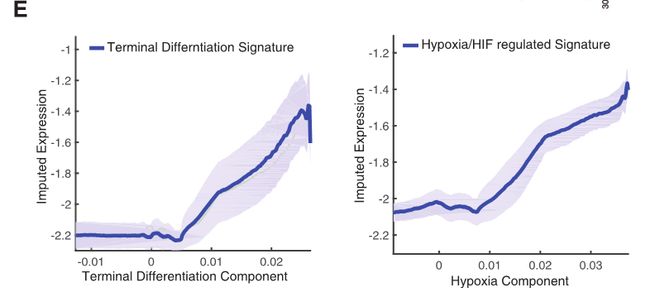
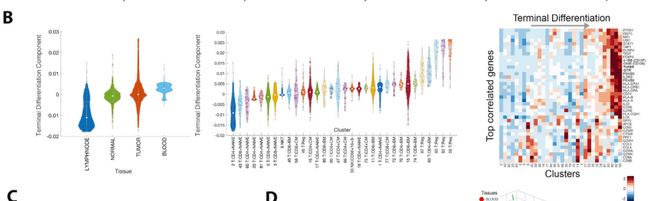
Fig 4E: 第二大信息组分— “终末分化”;第三大信息组分—“缺氧”。
与终末分化最相关的基因包括共刺激分子(CD2、GITR、OX40和4-1BB)以及共抑制受体(CTLA-4和TIGIT)(图S4B);此外,FOXP3、IL2RA和ENTPD1(CD39),TREG细胞的特征基因沿着终末分化组分轴,其表达量逐渐增加。
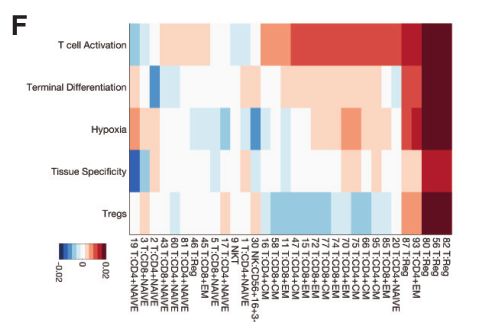
Fig 4F: 激活和终末分化成分最相关的基因有中度重叠(图4A、4C和S4B),与之前的单细胞研究一致。但也有一些重要的区别,包括上图列出的耗尽标记,并且集群的顺序在这两个组件上是不同的(图4F)。一些簇,特别是淋巴结T细胞(如簇16),表达的激活水平高于终末分化(T检验,P=0.0),这与非淋巴组织中T细胞衰竭/终末分化占优势是一致的。
基于以上讨论:将T细胞活化和终末分化成分一起可视化显示出显著的连续性,本质上代表了一个连续的轨迹(图4A)。因此,T细胞存在于一个广泛的激活连续体中,这意味着它们传统的分类为相对较少的离散激活或分化亚型可能会大大简化组织中T细胞群体的表型复杂性。
Part 07:不同的环境特征决定特异的瘤内T细胞簇
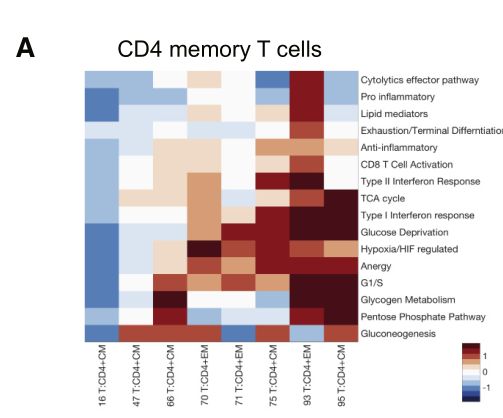
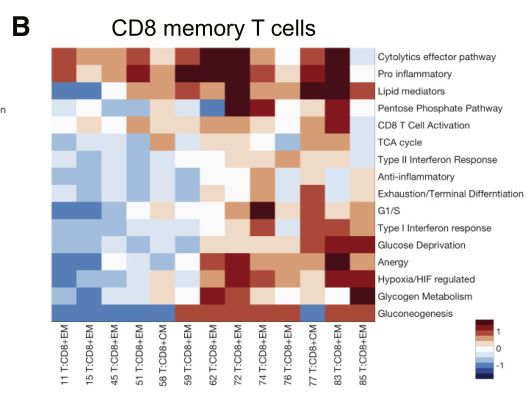
尽管簇沿着激活成分以连续的方式排列时,但是当考虑到与对不同环境刺激的反应相关联的标志组合时,每个簇看起来又是不同的。
Fig 5A:CD4效应记忆簇和中央记忆簇表现出与I型和II型干扰素应答(F检验,分别为P=1 X 10-54和0.008),缺氧(F检验,P=4 X 10-64),无能(anergy)(F检验,P=4 X 10-69)相关的基因表达水平的差异。
Fig 5B: CD8效应记忆簇和中央记忆簇在激活(F检验,P=2 x 10-114),促炎(F-试验,p=1 x 10-39)和细胞溶解效应途径(F-检验,P=6 x 10-32)相关基因的表达水平。
这些发现表明,肿瘤细胞可能暴露在不同程度的炎症,缺氧和营养缺乏条件。虽然许多这些反应(如激活或缺氧)单独代表表型连续性,但它们的组合可能导致更离散的状态。
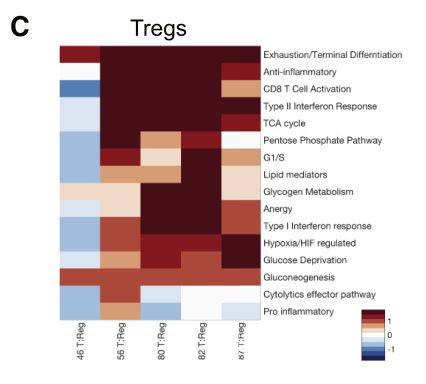
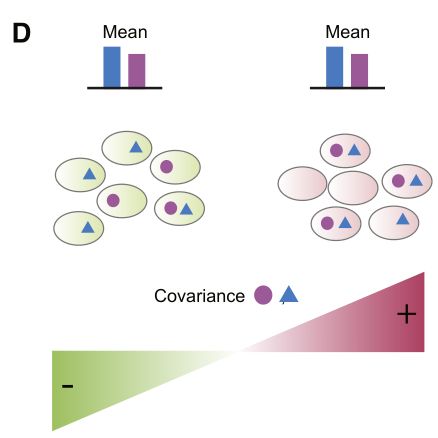
Fig 5C:与效应T细胞相比,大多数Treg细胞簇具有类似的抗炎、衰竭、缺氧和代谢基因集模式。
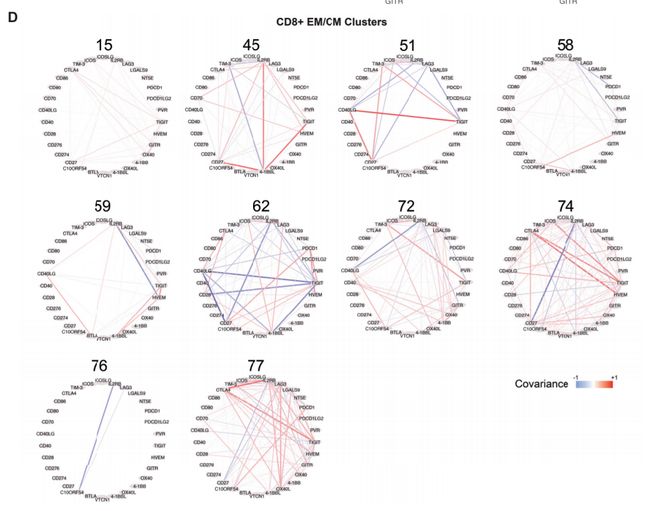
Fig 5D:协方差参数有助于推断聚类。
为了识别区分具有相似特征的Treg聚类,研究者研究区分Treg聚类的biscuit参数,发现在平均表达水平之上,簇之间的协方差参数变化显著。具体来说,两个标记基因在两个不同的簇中表现出相似的平均表达(例如,在两个簇中都高度表达),而这些簇在这些基因之间的协方差中表现出相反的迹象。这是因为基因通常在一个簇中的同一个细胞中共同表达(即正协方差),但在另一个簇中以互斥方式表达(即负协方差)。需要注意的是,聚类是根据14000多个基因的表达推断出来的,故两个特定基因之间的负协方差并不一定意味着亚聚类的存在。
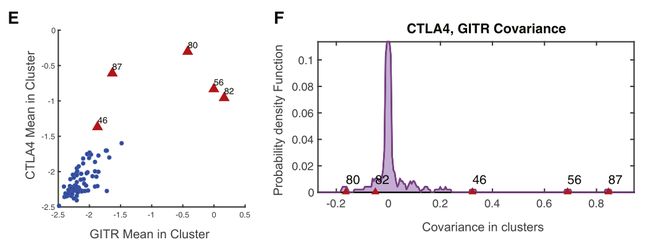
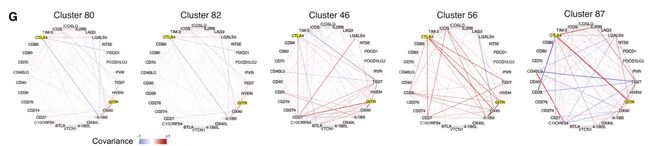
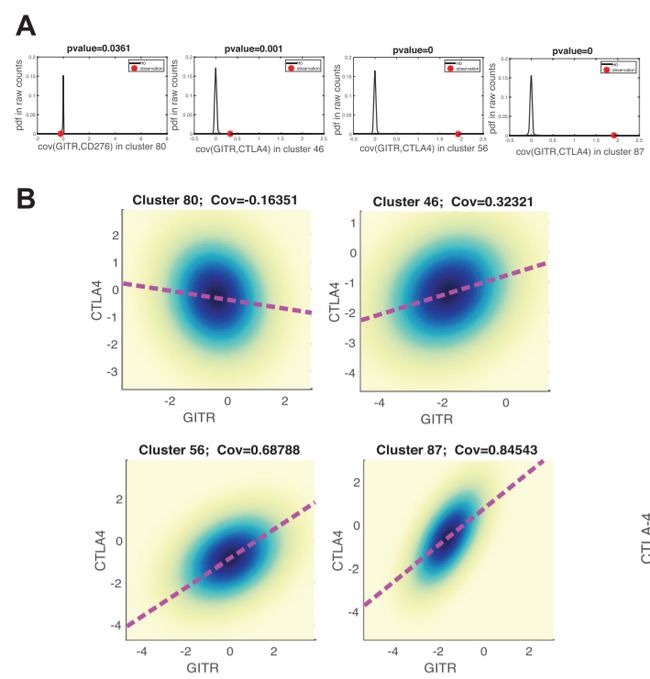
Fig 5E-5G;SF 5A-5B:典型的共抑制基因CTLA-4与其他机制上相关基因表现出丰富的协方差模式。CTLA-4与Treg的46、56和87簇中的TIGIT和共刺激受体GITR;与46和80簇中的CD27以及80簇中的共刺激受体ICOs有强烈的协同变化。
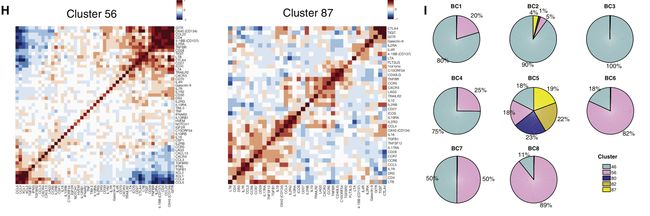
Fig 5H: 检查点受体之间的协方差模式通常在Treg簇中不同(图5G),其他重要的免疫基因表现出模块化协方差结构,表明在类似的功能模式中存在协同调节。
Fig 5I: 单个患者样本中观察到不同比例的Treg簇。因此在患者水平上也存在基因共表达的差异,并且大多数患者没有全部5种treg细胞亚型。
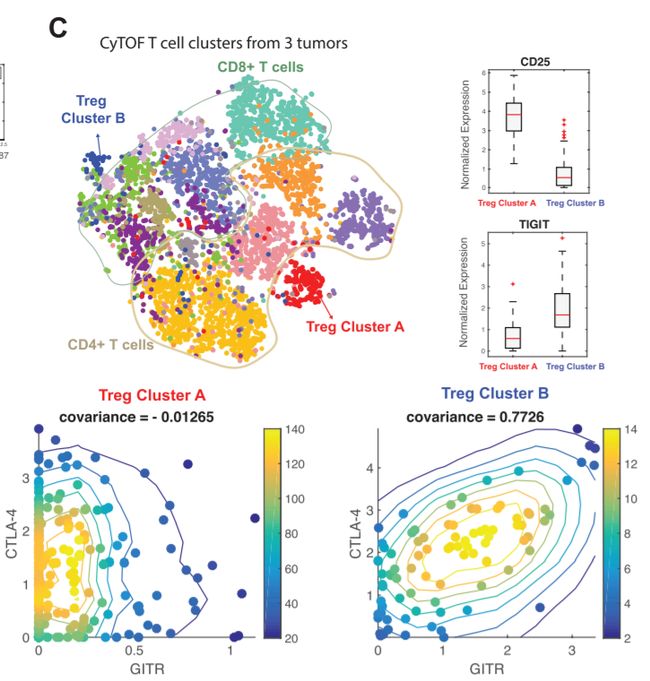
SF 5C-5D: 在另外3个乳腺肿瘤中观察到类似的GITR和CTLA-4模式。
方法:用CYTOF流式细胞术分析。
结果:2个Treg簇在协方差和差异表达基因方面与82和46簇相似(左图)。激活的T细胞簇的协方差模式丰富(右)。
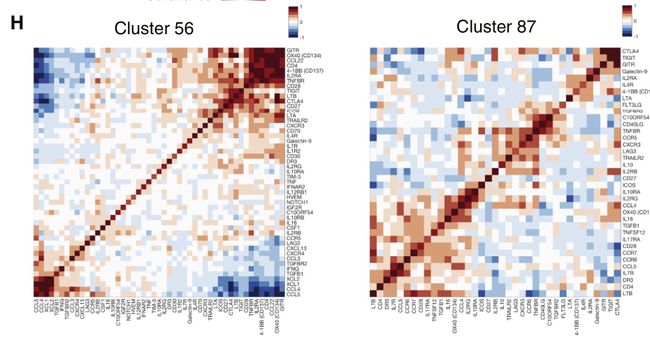
Fig 5H: 2个Treg簇免疫基因协方差值的热图显示了共变基因的不同模块。
故,基因的共变异在定义T细胞簇,特别是treg簇中起重要作用。
PART 08: 配对的单细胞RNA和TCR测序揭示单个T细胞克隆型的激活状态范围
肿瘤内T细胞活化的连续性的一个合理解释是暴露于不同的微环境中。一个非互斥的假设是,一系列TCR所提供的广泛信号强度可以导致T细胞激活的连续谱,从而掩盖过渡状态。支持后一种可能性,最近的一项研究表明,TCR转基因单克隆T细胞对同源肿瘤新抗原的识别导致激活的T细胞通过可逆的功能障碍中间状态有序地向不可逆的功能障碍终末状态发展
为了更深入地了解TCR序列多样性是否有助于T细胞活化谱和总体表型多样性的观察,研究者从另外3个乳腺癌患者中分离出27000个以上的CD3+T细胞,并进行单细胞RNA序列和成对V(D)J序列测定。这些数据允许同一个细胞将基因表达直接映射到TCR利用上。
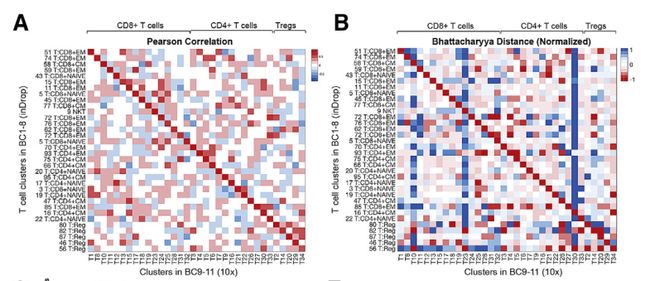
Fig 6A-6B: 因9个患者是在不同的单细胞平台上分析的,故转录组学数据进一步可以测试推断的簇对3个新患者的通用性。该分析显示,在10X平台生成的集合数据集上,使用biscuit识别的T细胞簇显示出与从Indrop数据集推断的T细胞簇接近一对一比对。
SF 6C: 该分析再现了T细胞激活轨迹上的连续梯度,类似于图4D所示。
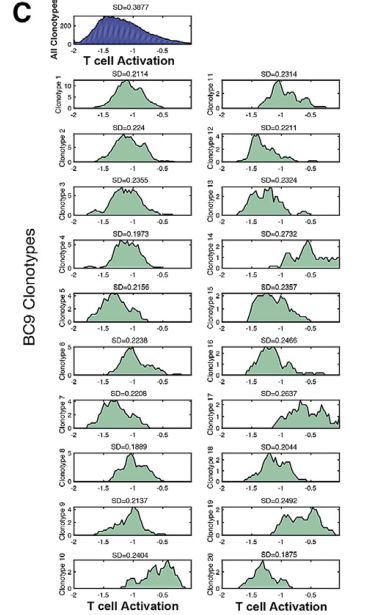
目的和方法:为了评估TCR利用在何种程度决定T细胞状态的连续型,研究者使用成对的单细胞RNA和TCR序列作图,并进行前100和前20频率最高克隆亚型的one-way ANOVA分析。
Fig 6C:每种克隆型在T细胞上的激活状态分布情况,每个TCR克隆亚型均分布在一个较广的T细胞激活状态范围。标准偏差(SD)表示TCR克隆亚型在不同状态中的离散情况,表明不同的克隆亚型表现出不同的平均激活水平。图S6E展示在BC10,11患者中与图6C同样的分析。图S6D展示不同克隆型的频率。

单因素方差分析的结果提示:如前100频率最高的BC9,对应52%,前100频率最高的TCR亚克隆仅能解释的变异为52%。其余解释亦是如此,代表TCR多样性并不是T细胞激活连续性的唯一驱动因素,相反可能还有其他因素的作用。
PART 09:T细胞的状态由TCR亚型决定
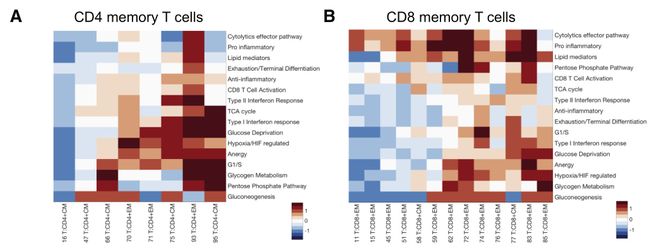
Fig 5A-B: 虽然免疫细胞变化的主要成分,如活化,显示出连续性,但T细胞簇根据其与环境刺激反应相关的特征的差异表达是可分离的。
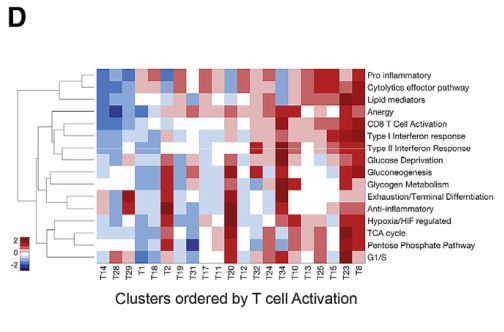
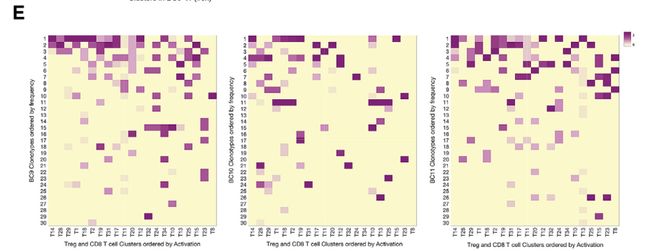
Fig 6D:10X技术分析的肿瘤组织驻留T细胞也有类似的趋势。
Fig 6E:当与TCR克隆型联合分析时,发现每个簇实际上由克隆型的不同组合子集组成。这一观察结果进一步支持TCR表达谱可部分解释细胞簇的独特性。
对于含同一TCR克隆亚型的T细胞聚类,展现出相似的激活水平,也就是说拥有同一类型TCR亚型的T细胞聚类在微环境刺激中基因表达标志相似。例如BC9中,T11和T12聚类具有克隆亚型9,他们所有的表达标志的表达水平相似;如BC9中,共有克隆亚型16的两个T细胞聚类(20-34)展现出相似水平的耗竭标志,共有克隆亚型20的聚类(31-34)展现出相似的无能和糖异生水平;BC10中,共有克隆亚型21的聚类展现出相似的高水平的G1/S期标志,以及低水平的耗竭、缺氧、TCA循环和抗炎症标志(t检验,p<0.1)。
Fig 6F:对BC9-11肿瘤T细胞的标准化scRNA-seq数据进行t-SNE投射,T细胞活化标志和肿瘤样本染色(左); biscuit簇(中间顶部); 以及通过配对TCR测序鉴定出的每个肿瘤的显性克隆型,投影在相同的坐标上(右)。
发现每种克隆型仅存在于少数相关簇中,与亚簇相似程度高,故在t-SNE投影中占据了狭窄区域。
PART 10: 激活和分化解释了瘤内骨髓源性细胞的变异
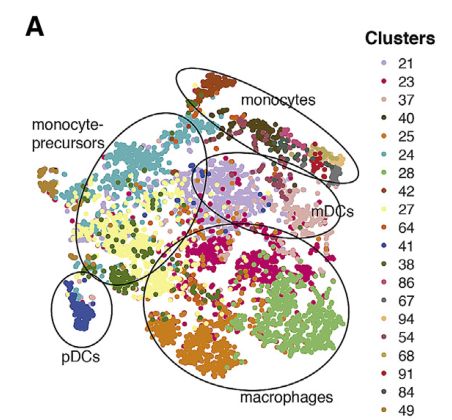
Fig 7A: 尽管髓样细胞是肿瘤微环境的关键组成部分,但它们的异质性及其对肿瘤进展的影响仍然不足以表征。对BC1-8中发现的单核细胞簇的广泛调查表明在这些主要细胞类型中存在未探索的亚结构。
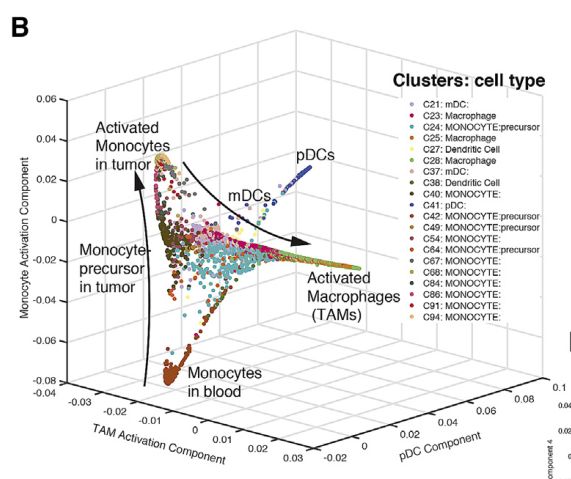
Fig 7B: 与T细胞一样,使用扩展映射来评估髓细胞的变异性,不包括嗜中性粒细胞和肥大细胞,因为它们形成了更多不同的簇。
结果展示:第一分支几乎全部包含来自3个簇(23、25和28)的瘤内巨噬细胞(TAM)。接下来的两个组成部分捕获了从血液单核细胞到肿瘤内单核细胞的渐进轨迹;具有2个离散状态的附加组分将浆细胞样DC(pDC)与其他单核细胞簇区分开。
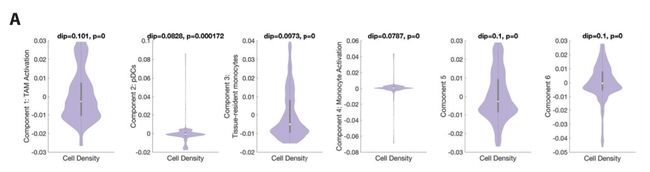
SF 7A: 扩展映射分析揭示了四个主要分支,它们比T细胞显示出更多不同的细胞状态(图S7A)
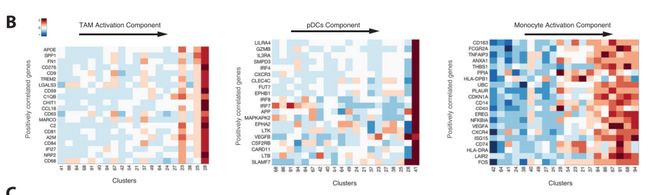
SF 7B:第一个主要分支-TAM激活组分。
- 最相关的基因包括APOE,CD68,TREM2和CHIT1,可能反映募集的或组织驻留的巨噬细胞的分化和激活。
- 与“交替激活的”(M2)巨噬细胞相关的基因的表达,包括清道夫受体MARCO,促血管生成受体NRP2和抑制性分子B7-H3(CD276)沿该分支增加。
-
与“经典激活”(M1)巨噬细胞相关的免疫刺激基因(包括趋化因子CCL3(MIP-1a))沿分支增加。
 54.png
54.png
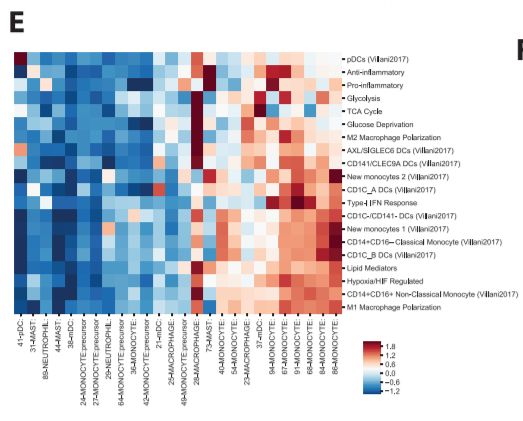
SF 7E: TAM的所有3个簇,特别是23和28簇,都属于典型M2标志表达高的单核细胞簇但M1标志同样很高。
 55.png
55.png
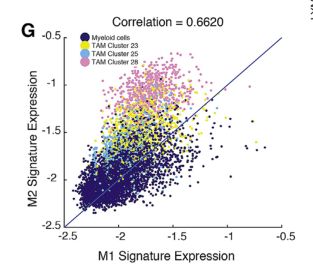
Fig 7G:髓样群体中的M1和M2基因标志呈正相关。
这与其他肿瘤类型的最新发现一致。这些发现证明,无论是离散状态还是沿极化轨迹谱状态的极化模型,肿瘤微环境中的巨噬细胞活化与极化不符。
 56.png
56.png
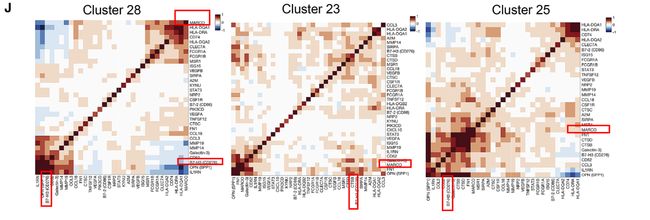
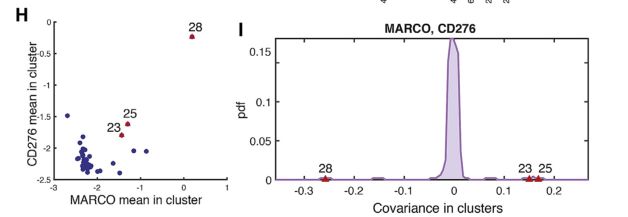
Fig 7H-J: 协方差参数分析:协方差参数是区分3个TAM亚聚类。
如:探究2种M2型标记物MARCO和CD276的共表达——尽管TAM簇均表达高水平的两个基因,但它们在簇23和25中呈正相关,而在簇28中呈负相关
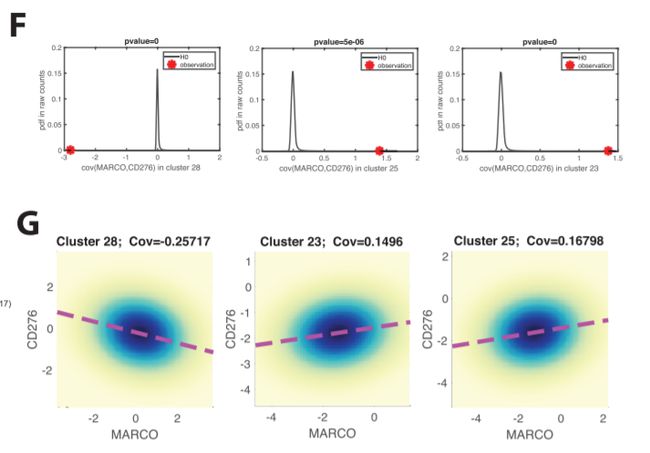
SF 7F:原始数据中:在28,25,23TAM簇中仍有不同的协方差模式,与标准化后的结果一致。
故协方差是数据本身的差异产生,不是建模的产物。
这些结果突出了共表达模式在定义髓样细胞状态中的重要性。
最后,我们再回顾全文主要内容: