PS:我只是个搬运工
文献中的热图的解读:
http://www.360doc.com/content/18/0316/15/51841481_737529918.shtml
热图是对实验数据分布情况进行分析的直观可视化方法,可以用来进行实验数据的质量控制和差异数据的具像化展示,还可以对数据和样品进行聚类,观测样品质量。
它有多种形式,但基本的元素却是通用的。例如下图,就是一副组学研究中热图的常用绘制模式,每个小方格表示每个基因,其颜色表示该基因表达量大小,表达量越大颜色越深(红色为上调,绿色为下调)。每行表示每个基因在不同样本中的表达量情况,每列表示每个样品中所有基因的表达量情况。上方树形图表示对来自不同实验分组的不同样品的聚类分析结果,左侧树状图表示对来自不同样本的不同基因的聚类分析结果:
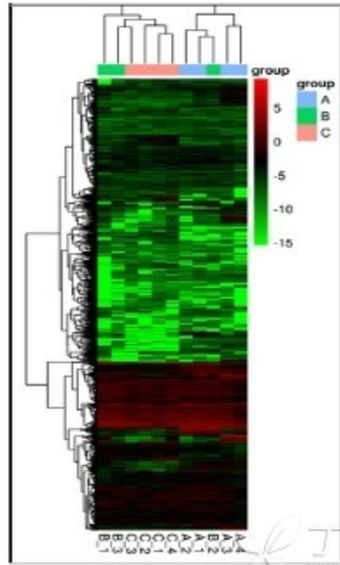
在文章中,热图通常有两大作用:数据质量控制和直观展示重点研究对象的差异变化情况。
首先,我们来说说热图在数据质量控制(质控)过程中的应用。如下图,通过观察上方树形图(对列的聚类分析)大家可以明显的看到,两个实验分组中的Experiment组,其基因的总体表达模式与作为对照的control组存在很大的差异:在对照组中,多数基因都呈现极大下调的表达模式(绿色条带),而实验组则正好相反,多数基因为上调模式。属于对照组的三个样本(C1、C4和C5)的表达模式相似,属于实验组的三个样本(E1、E4和E5)的表达模式相似。这证明无论是对照组还是实验组,各自的样本重复性都是很好的,至少可以证明其在实验处理上是不存在大的失误的,得出的数据也是可信的、可靠的、符合逻辑的。而一旦出现同一组的某一个或某几个样本的表达模式与本组内其他样本表达模式迥异的情况,则需要小心调查前期实验是否存在问题了。
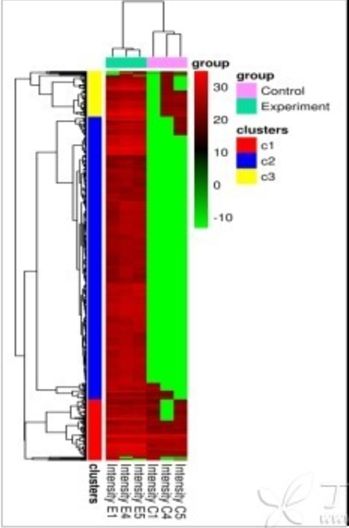
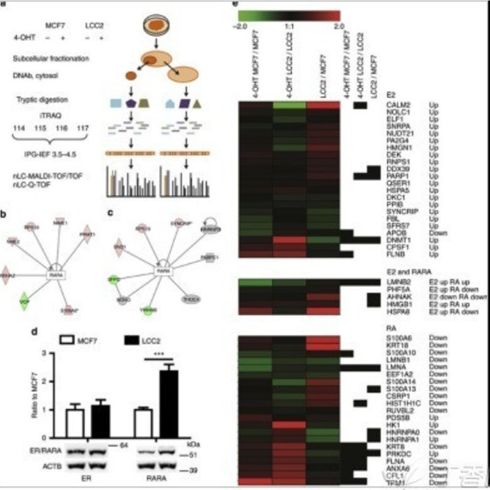
热图在文章中的另一大作用就是直观展示重点研究对象的表达量数据差异变化情况。通过上文的介绍,大家可能已经发现了一个问题,那就是一次实验中检测到的基因或蛋白往往成干上万,导致一副全局性的热图的行数(基因或蛋白数)也相应地十分庞大,使得在一副图片大小的篇幅内,代表单个基因或蛋白的每一个小方格的信息(如基因名和表达量等)几乎不可能被肉眼所识别。所以类似的用整个数据集画出的热图往往只能用于数据的整体质控。而要能够向读者清晰展示自己所研究的某一批基因或蛋白的数据分布与变化情况,就应当如下图所示(原文链接http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3759040/),把自己的研究对象从数据集中找出来再绘制热图(作为重点关注的几个标志性的基因在右侧以黑色标注):
那么热图应该如何绘制呢?
https://blog.csdn.net/lalaxumelala/article/details/86022722?utm_medium=distribute.pc_relevant.none-task-blog-baidulandingword-8&spm=1001.2101.3001.4242
首先查看R当中的heatmap说明书:
格式:
heatmap(x, Rowv = NULL, Colv =if(symm)"Rowv" else NULL,
distfun = dist, hclustfun =hclust,
reorderfun = function(d, w)reorder(d, w),
add.expr, symm = FALSE, revC =identical(Colv, "Rowv"),
scale = c("row","column", "none"), na.rm = TRUE,
margins = c(5, 5), ColSideColors,RowSideColors,
cexRow = 0.2 + 1/log10(nr),cexCol = 0.2 + 1/log10(nc),
labRow = NULL, labCol = NULL,main = NULL,
xlab = NULL, ylab = NULL,
keep.dendro = FALSE, verbose =getOption("verbose"), ...)
参数:
x
numeric matrix of
the values to be plotted.要绘制的值的数字矩阵。
Rowv
determines if and
how the row dendrogram should be computed and reordered. Either a dendrogram or
a vector of values used to reorder the row dendrogram or NA to suppress any row
dendrogram (and reordering) or by default, NULL, see ‘Details’ below.确定是否以及如何计算和重新排序行树状图。树状图或用于对行树状图进行重新排序的值向量,或用于抑制任何行树状图(和重新排序)的NA,默认情况下为NULL,请参阅下面的“详细信息”。
Colv
determines if and
how the column dendrogram should be reordered. Has the same options as the Rowv
argument above and additionally when x is a square matrix, Colv =
"Rowv" means that columns should be treated identically to the rows
(and so if there is to be no row dendrogram there will not be a column one
either).确定是否以及如何对列树状图重新排序。与上面的Rowv参数具有相同的选项,此外,当x是方阵时,Colv = "Rowv"意味着列应该与行相同(因此,如果没有行树状图,也不会有列树状图)。
distfun
function used to
compute the distance (dissimilarity) between both rows and columns. Defaults to
dist.函数,用于计算行和列之间的距离(差异)。默认为dist。
hclustfun
function used to
compute the hierarchical clustering when Rowv or Colv are not dendrograms.
Defaults to hclust. Should take as argument a result of distfun and return an
object to which as.dendrogram can be applied.当Rowv或Colv不是树状图时,用于计算层次聚类的函数。默认为hclust。应该以distfun的结果作为参数,并返回一个可应用as.dendrogram的对象。
reorderfun
function(d, w) of
dendrogram and weights for reordering the row and column dendrograms. The
default uses reorder.dendrogram.树状图的函数(d, w)和用于对行、列树状图重新排序的权重。默认使用reorder.dendrogram。
add.expr
expression that
will be evaluated after the call to image. Can be used to add components to the
plot.在调用image之后计算的表达式。可用于向绘图添加组件。
symm
logical indicating
if x should be treated symmetrically; can only be true when x is a square
matrix.逻辑指示x是否应被对称对待;只有当x是方阵时才成立。
revC
logical indicating
if the column order should be reversed for plotting, such that e.g., for the
symmetric case, the symmetry axis is as usual.逻辑指示在绘图时列的顺序是否应该颠倒,例如,在对称情况下,对称轴是正常的。
scale
character indicating
if the values should be centered and scaled in either the row direction or the
column direction, or none. The default is "row" if symm false, and
"none" otherwise.字符,指示值是否应居中并按行或列方向缩放,或不按行或列缩放。如果symm为false,默认为“row”,否则为“none”。
na.rm
logical indicating
whether NA's should be removed.逻辑指示是否应该移除NA。
margins
numeric vector of
length 2 containing the margins (see par(mar = *)) for column and row names,
respectively.长度为2的数字向量,分别包含列和行名称的边距(参见par(mar = *))。
ColSideColors
(optional)
character vector of length ncol(x) containing the color names for a horizontal
side bar that may be used to annotate the columns of x.(可选)长度ncol(x)的字符向量,包含水平侧栏的颜色名称,可用于标注x的列。
RowSideColors
(optional)
character vector of length nrow(x) containing the color names for a vertical
side bar that may be used to annotate the rows of x.(可选)长度为nrow(x)的字符向量,包含用于注释x行的垂直侧栏的颜色名称。
cexRow, cexCol
positive numbers,
used as cex.axis in for the row or column axis labeling. The defaults currently
only use number of rows or columns, respectively.正数,用cex表示。轴在用于行或列轴标记。目前,默认值仅分别使用行数或列数。
labRow, labCol
character vectors
with row and column labels to use; these default to rownames(x) or colnames(x),
respectively.要使用的带有行和列标签的字符向量;它们分别默认为rownames(x)或colnames(x)。
main, xlab, ylab
main, x- and
y-axis titles; defaults to none.主、x轴和y轴标题;默认为没有。
keep.dendro
logical indicating
if the dendrogram(s) should be kept as part of the result (when Rowv and/or
Colv are not NA).逻辑指示树状图是否应该作为结果的一部分(当Rowv和/或Colv不是NA时)。
verbose
logical indicating
if information should be printed.
逻辑指示是否应该打印信息。
其他基础设置
main 图的名字
file 要保存图的名字
color 表示颜色,赋值渐变颜色调色板colorRampPalette属性,选择“绿,黑,红”渐变,分为100个等级,,例:color = colorRampPalette(c(“navy”, “white”, “firebrick3”))(102)
sclae 表示值均一化的方向,或者按照行或列,或者没有,值可以是"row", “column” 或者"none"
margins 表示页边空白的大小
fointsize 表示每一行的字体大小
聚类相关设置
cluster_cols 表示进行列的聚类,值可以是FALSE或TRUE
cluster_row 同上,是否进行行的聚类
treeheight_row 设置row方向的聚类树高
treeheight_col 设置col方向的聚类树高
clustering_distance_row 表示行距离度量的方法
clustering_distance_cols 同上,表示列距离度量的方法
clustering_method 表示聚类方法,值可以是hclust的任何一种,如"ward.D",“single”, “complete”(默认), “average”, “mcquitty”, “median”, “centroid”, “ward.D2”
legend设置
legend TRUE或者FALSE,表示是否显示图例
legend_breaks 设置图例的断点,格式:vector
legend_labels legend_breaks对应的标签 例:legend_breaks = -1:4, legend_labels = c(“0”,“1e-4”, “1e-3”, “1e-2”, “1e-1”, “1”)
单元格设置
border_color 表示热图上单元格边框的颜色,如果不绘制边框,则使用NA
cellheight 表示每个单元格的高度
cellwidth 表示每个单元格的宽度
单元格中的数值显示:
display_numbers 表示是否将数值显示在热图的格子中,如果这是一个矩阵(与原始矩阵具有相同的尺寸),则显示矩阵的内容而不是原始值。
fontsize 表示热图中字体显示的大小
number_format 设置显示数值的格式,较常用的有"%.2f"(保留小数点后两位),"%.1e"(科学计数法显示,保留小数点后一位)
number_color 设置显示内容的颜色
热图分割设置
cutree_rows 基于层次聚类(使用cutree)划分行的簇数(如果未聚集行,则忽略参数)
cutree_cols 基于层次聚类(使用cutree)划分列的簇数
annotation相关设置
annotation_row 行的分组信息,需要使用相应的行名称来匹配数据和注释中的行,注意之后颜色设置会考虑离散值还是连续值,格式要求为数据框
annotation_col 同上,列的分组信息
annotation_colors 用于手动指定annotation_row和annotation_col track颜色的列表。
annotation_names_row boolean值,显示是否应绘制行注释track的名称。
[if !supportLists]· [endif]annotation_names_col 同上,显示是否应绘制列注释track的名称
下面尝试进行绘制:
# 测试数据
a=c(12,14,17,11,16)
b=c(4,20,15,11,9)
c=c(5,7,19,8,18)
d=c(15,13,11,17,16)
e=c(12,19,16,7,9)
A=cbind(a,b,c,d,e)
B=rbind(a,b,c,d,e)
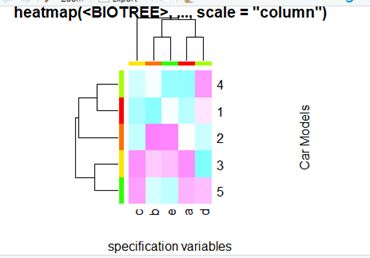
require(graphics); require(grDevices)
x <-as.matrix(A)
rc <- rainbow(nrow(x), start = 0, end = .3)
cc <- rainbow(ncol(x), start = 0, end = .3)
hv <- heatmap(x, col = cm.colors(256), scale ="column",
RowSideColors= rc, ColSideColors = cc, margins = c(5,10),
xlab = "specification variables", ylab = "Car Models",
main = "heatmap(, ..., scale =\"column\")")
utils::str(hv)
关于其他设置还需要再熟悉一遍
下面跟随网友的帖子跑一遍过程
https://www.jianshu.com/p/718b22bb1148
1. 安装pheatmap包
install.packages("pheatmap")
library(pheatmap)
2. 创建测试矩阵
test = matrix(rnorm(200), 20, 10) #test为一个20*10的矩阵,200个元素满足参数为0和1的正态分布
test[1:10, seq(1, 10, 2)] = test[1:10, seq(1, 10, 2)] + 3
test[11:20, seq(2, 10, 2)] = test[11:20, seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20, seq(2, 10, 2)] + 4
colnames(test) = paste("Test", 1:10, sep = "") #
定义列名,注意paste的用法
rownames(test) = paste("Gene", 1:20, sep = "") #
定义行名
rownames(test) = paste("Gene", 1:20, sep = "") #定义行名
3. 画个热图
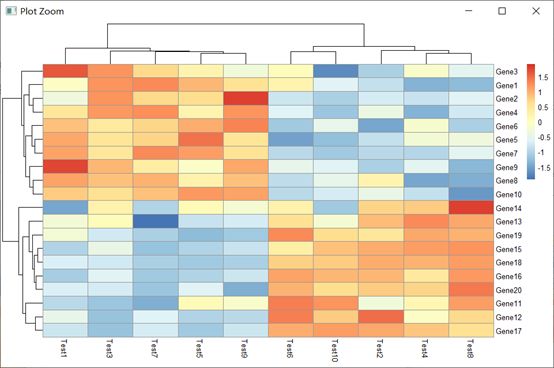
3.1 pheatmap(test)
基本用法,根据“2. 创建测试矩阵”中的局部赋值运算可以得到明显的分区。
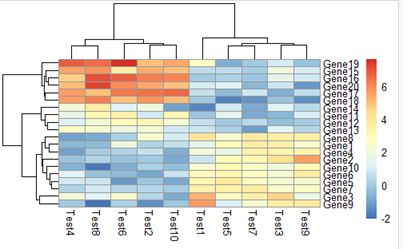
3.2 pheatmap(test, kmeans_k = 3)
将行聚为几类
kmeans是一种聚类算法,详见https://www.cnblogs.com/bourneli/p/3645049.html
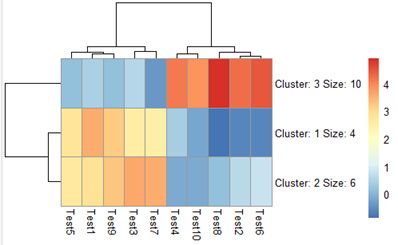
3.3 pheatmap(test, scale = "row")
标准化
为什么要标准化? 原始数据中,每个基因表达变化范围对应的数值大小不同,导致图片中色彩变化难以显示基因在不同样本中的变化趋势,可以对基因在每个样本中基因表达数据进行标准化,使其数值在一定范围内,从而实现热图的优化,而控制参数为scale,对基因(行,row)进行处理
可以与第一张图比较一下,例如Gene3这一行,在这张图中可以看出样本之间的差异明显了许多。
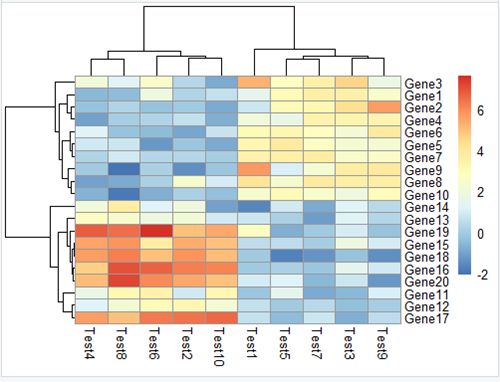
3.4 pheatmap(test, clustering_distance_rows =
"correlation")
聚类线长度优化,可能不一样的算法有不一样的枝长。
clustering_distance_cols同理。
3.5 pheatmap(test, color =
colorRampPalette(c("navy", "white",
"firebrick3"))(10))
设置颜色,后面括号里的数字表示梯度,10就是将这三种颜色设置为10个梯度

3.6 pheatmap(test, cluster_row = FALSE)
是否显示行的聚类,cluster_col同理
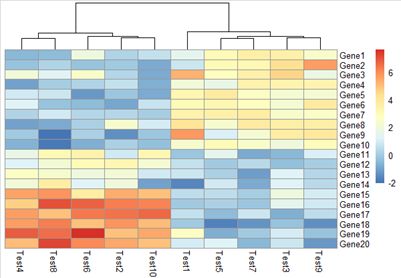
3.7 pheatmap(test, legend = FALSE)
是否显示图例
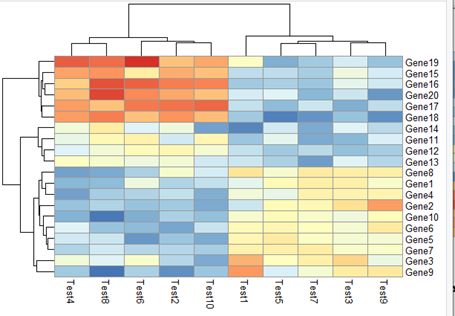
4. 显示色块的数值或文本
基本用法:pheatmap(test,display_numbers = TRUE)
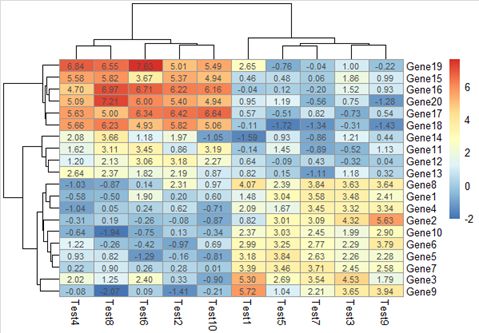
此外还可添加如下参数
number_format = "%.3e"表示保留3位小数,且用科学计数法显示
number_format = "%.3f"表示保留3位小数,用小数显示
display_numbers除了赋布尔值,还能赋矩阵(其维度与原矩阵相同),此时可以人为添加文本(有点像R画图的图层叠加)。
pheatmap(test, display_numbers = matrix(ifelse(test > 5, "*",
""), 20,10))

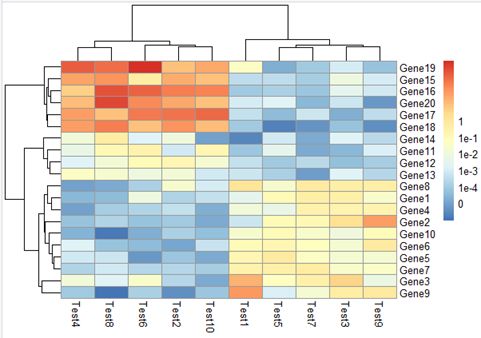
legend_breaks设置图例的显示范围,间隔为1;legend_labels重写刻度的标签, 需与legend_breaks同时使用。
pheatmap(test, legend_breaks = -1:4,legend_labels = c("0","1e-4", "1e-3","1e-2", "1e-1", "1"))
5. 调整色块或文本大小并导出文件
pheatmap(test, cellwidth = 15, cellheight = 12,main = "Example heatmap", fontsize = 8, filename ="test.pdf")
dev.off()
这五个参数分别表示:
色块的宽度、色块的高度、标题、行列名及图例字体的大小、保存为当前工作目录下的图片的文件名

6. 行列注释
对于每一行每一列都添加一些注释信息,本质还是"分类"。
annotation_col = data.frame(
CellType =factor(rep(c("CT1", "CT2"), 5)),
Time = 1:5
) #
注意rep()的用法;为什么要定义为因子;R可以自动补全Time变量
rownames(annotation_col) = paste("Test", 1:10, sep = "")
annotation_row =data.frame(
GeneClass =factor(rep(c("Path1", "Path2", "Path3"), c(10, 4,6)))
)
rownames(annotation_row) = paste("Gene", 1:20, sep = "")
> annotation_col
CellType Time
Test1 CT1 1
Test2 CT2 2
Test3 CT1 3
Test4 CT2 4
Test5 CT1 5
> annotation_row
GeneClass
Gene1 Path1
Gene2 Path1
Gene3 Path1
Gene4 Path1
Gene5 Path1
pheatmap(test, annotation_col = annotation_col, annotation_row =
annotation_row)

自定义注释色块的颜色
ann_colors = list(
Time = c("white","firebrick"),
CellType = c(CT1 = "#1B9E77",CT2 = "#D95F02"),
GeneClass = c(Path1 ="#7570B3", Path2 = "#E7298A", Path3 = "#66A61E")
) #
注意ann_colors是列表
pheatmap(test, annotation_col = annotation_col,annotation_row = annotation_row,
annotation_colors = ann_colors)
7. 列名的文本角度调整
angle_col = "45",文本与从左向右水平线的夹角,只能是“270”, “0”, “45”, “90”, “315”这几个值。
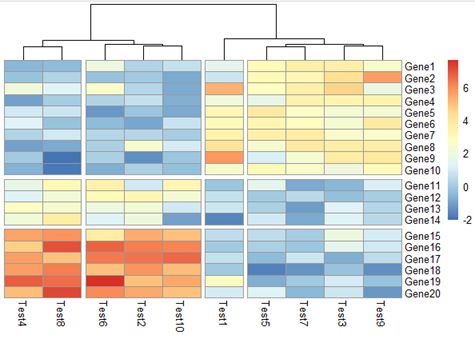
8. 切分热图
pheatmap(test, cluster_rows = F, gaps_row = c(10,14), cluster_cols = T,
cutree_col = 4)
gaps_row有效的前提是cluster_rows = F;cutree_col有效的前提是cluster_cols = T
效果图如下:
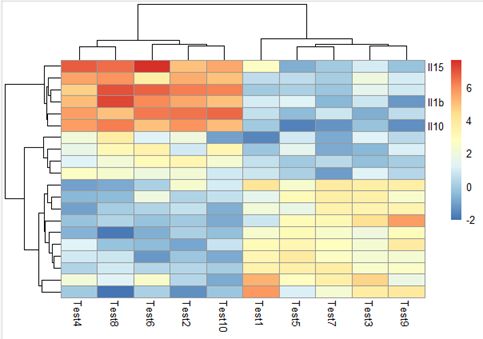
9. 自定义显示哪些行列的名字
labels_row = c("", "","", "", "", "", "","", "", "", "", "","", "", "",
"","", "Il10", "Il15", "Il1b")
pheatmap(test, labels_row = labels_row)
更多的细节就需要在使用的时候一点点调试了
参考资料:
http://www.360doc.com/content/18/0316/15/51841481_737529918.shtml
https://www.cnblogs.com/bourneli/p/3645049.html
https://www.jianshu.com/p/718b22bb1148