- 数据库
小胡123
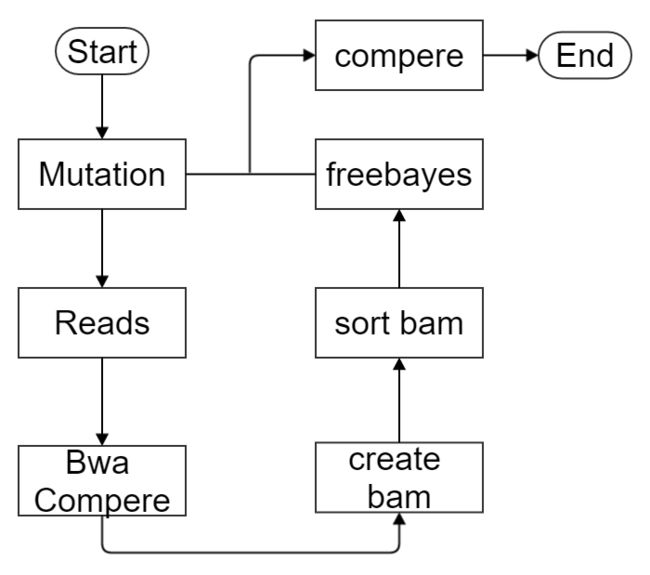
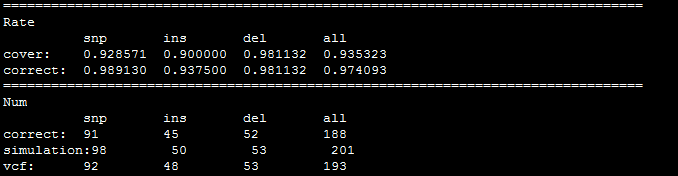
数据库长期保存在计算机的存储设备上,按照一定规则组织起来,可被各种用户或应用共享的数据集合数据库系统(DBS)采用数据库技术的计算机系统,由数据库,数据库管理系统,数据库管理员,硬件平台,软件平台构成数据库管理系统(DBMS)是操作和管理数据库的软件,用于建立,使用和维护数据库,对数据库进行统一管理和控制提供的功能:数据定义语言(DDL)数据操作语言(DML)数据控制语言(DCL)数据存储方式:l
- junit mockito_如何学习Java中的单元测试:JUnit和Mockito课程
dfsgwe1231
单元测试编程语言python人工智能java
junitmockito大家好,今天我将讨论JUnit和单元测试,这是任何软件开发人员的关键技能之一。您可能已经知道JUnit和Mockito是Java应用程序中最受欢迎的两个测试库,并且几乎在每个Java应用程序类路径中都可以找到它们。我经常与Java开发人员见面并一起工作,这些Java开发人员非常了解Java但还没有编写单个单元测试。当我问他们为什么不编写单元测试时,他们提出了许多借口,例如他
- 9分钟了解彦祖文化app不能出金黑幕曝光!真相让人震惊!
最新曝光36
希望看到这篇文章的人可以及时止损;请及时联系为你提供解决方案,要想讨回损害资产务必阅读以下内容。1.彦祖文化APP在平台不能提现怎么办?2.彦祖文化APP这个软件靠谱可信吗?3.彦祖文化APP在软件做任务被骗?4.彦祖文化APP软件app无法登录?5.彦祖文化APP平台是真的吗?6.彦祖文化APP被骗无法提现,操作失误!7.彦祖文化APP平台是騙局吗?8.彦祖文化APP被骗无法出金如何维护自己的合
- 25岁从零开始学习平面设计,会不会太晚?
93091cdf8ebb
很多新手小白想学平面设计,但是苦于不知从何处入门、怎么样去学。究竟怎样系统学习平面设计?今天就来谈谈平面设计系统学习的方法。更多学习设计内容关注V公众号广告设计之站了解平面设计平面设计是以“视觉”作为沟通和表现的方式,通过文字、图片等媒介有机结合,借此表达视觉上的讯息。平面设计的分类有很多,如:名片设计、标志设计、字体设计、VI视觉形象设计等等。所以设计师要掌握字体排印、视觉艺术、版面、电脑软件等
- Day9: OpenCV学习(一)—— 图像基础
系列文章目录上一篇:Day8:Python工程化——模块、包文章目录系列文章目录前言一、安装和导入1.安装二、图像认识1.图像2.图像分类三、基础图像操作1.图像读取2.图像显示3.图像裁剪4.图形尺寸修改5.图像保存6.图像绘制7.视频捕获即显示总结前言OpenCV(OpenSourceComputerVisionLibrary)是一个开源的计算机视觉和机器学习软件库。由一系列C++类和函数构成
- Ubuntu的apt、apt-get和snap闲聊(2025年3月28日)
为什么Ubuntu中有了APT、APT-GET还要加上Snap?在Ubuntu系统中,软件管理工具的多样性(如APT、APT-GET和Snap)常常让人疑惑:既然已经有了成熟的APT和APT-GET,为什么还要引入Snap?本文将从不同角度解析这一问题,探讨Snap的独特价值及其与传统工具的共存意义。这份笔记适用于Linux用户、开发者以及对软件生态感兴趣的读者,内容将随技术演进保持更新。QA:解
- API开发全攻略:从入门到精通的企业级API架构与实战
Android洋芋
架构API设计RESTfulAPI微服务架构实战案例
简介API开发已成为现代软件架构的核心能力,掌握API设计与实现技术能显著提升开发效率和系统可扩展性。本文将从零开始,全面解析API的基础概念、架构设计、安全认证、性能优化等关键技术点,并提供完整的Python和Go语言代码实战示例,帮助开发者构建高性能、可扩展的企业级API系统。本文旨在为初学者和进阶开发者提供一份全面的API开发指南。内容涵盖API的基础概念、类型分类、架构设计、安全认证、性能
- 电子电气架构 --- 从软件质量看组织转型路径
汽车电子实验室
电子电器架构开发流程EV(电动汽车)常规知识必备架构电子电气架构电气电子架构开发的应对策略开发语言ECU刷写与busoff原则
我是穿拖鞋的汉子,魔都中坚持长期主义的汽车电子工程师。老规矩,分享一段喜欢的文字,避免自己成为高知识低文化的工程师:简单,单纯,喜欢独处,独来独往,不易合同频过着接地气的生活,除了生存温饱问题之外,没有什么过多的欲望,表面看起来很高冷,内心热情,如果你身边有这样灵性的人,一定要好好珍惜他们眼中有神有光,干净,给人感觉很舒服,有超强的感知能力有形的无形的感知力很强,能感知人的内心变化喜欢独处,好静,
- 电子电气架构 --- 汽车软件全生命周期
汽车电子实验室
电子电器架构开发流程车载电子电气架构架构汽车电气电子架构开发的应对策略开发语言ECU刷写与busoff原则电子电气架构
我是穿拖鞋的汉子,魔都中坚持长期主义的汽车电子工程师。老规矩,分享一段喜欢的文字,避免自己成为高知识低文化的工程师:简单,单纯,喜欢独处,独来独往,不易合同频过着接地气的生活,除了生存温饱问题之外,没有什么过多的欲望,表面看起来很高冷,内心热情,如果你身边有这样灵性的人,一定要好好珍惜他们眼中有神有光,干净,给人感觉很舒服,有超强的感知能力有形的无形的感知力很强,能感知人的内心变化喜欢独处,好静,
- 01-C语言:第01天笔记
Star在努力
c语言笔记开发语言
C语言:第1天笔记内容提要C语言概述数据类型常量变量C语言概述计算机基础计算机的组成计算机组成计算机:能进行计算以及逻辑处理的设备硬件:组成计算机的物理部件。(内存条、CPU、硬盘…)开发中对于硬件的认知:硬件包括电子设备、单片机、集成电路和嵌入式系统。软件:计算机中运行的程序和数据。开发中对于软件的认知:软件分为系统软件(OS)、应用软件和编程工具(编译器)计算机的六大部件中央处理器(CPU):
- 海淘平台哪个好 海淘app正品靠谱排行榜
氧惠评测
海淘软件让你可以在线选购来自全球各地的好物,海淘app有众多知名品牌入驻,保证正品,售后服务有保障,买的放心,海淘买的多省的多,直邮,正品产品包邮送到家。购物、看电影、点外卖、用氧惠APP!更优惠!氧惠(全网优惠上氧惠)——是与以往完全不同的抖客+淘客app!2023全新模式,我的直推也会放到你下面,送1:1超级补贴(邀请好友自购多少,你就推广得多少,非常厉害),欢迎各位团队长体验!也期待你的加入
- 如何打造个人品牌,从ID到IP
齐帆齐
如何打造个人品牌,从ID到IPID(IdentityDocument),是身份证标识号、账号、唯一编码、专属号码、工业设计、国家简称、法律词汇、通用账户、译码器、软件公司等各类专有词汇的缩写。我们的微信号,我们的任何平台注册号都有ID,我的ID就是我的首拼。有的人用首拼加上年份的名字,这是我们取ID的一个小窍门,让别人更方便地找到我们。从ID到IP,你要做好每一个细节,还要长期输出内容,尽可能在各
- 京东返利软件排行榜前十名,10款最热门的京东返利APP排行榜
古楼
在网购盛行的时代,优惠券和返利成为了吸引用户的重要手段。为了让广大消费者更好地享受到购物返利的好处,本文为您推荐了2024年十大热门的京东返利APP,帮助您在享受优惠的同时,还能获得额外返利。月入十万必看!都在挣钱!推荐几个月入几千到几万的靠谱副业项目!(公众号:善士思维笔记)【高省】APP(高佣金领导者)是一个自用省钱佣金高,分享推广赚钱多的平台,2000万用户信赖的四年老平台,稳定可靠。高省A
- 基于定制开发开源AI智能名片S2B2C商城小程序源码的搜索框个性化推荐机制研究
摘要:本文聚焦于定制开发开源AI智能名片S2B2C商城小程序源码场景下的搜索框个性化推荐机制。通过分析搜索框作为信息流槽位的产品形态特性,结合开源AI大模型与S2B2C模式的技术融合优势,提出基于用户强兴趣/即时兴趣的动态推荐策略。研究揭示了定制化开发在破解传统搜索框静态局限中的关键作用,并通过实证案例验证了该机制对提升用户转化率与平台GMV的显著效果,为新零售场景下的智能推荐系统设计提供了理论依
- ANSYS 2025 R1软件下载及安装教程|附安装文件
仰望天空—永强
嵌入式硬件硬件工程智能硬件硬件架构数学建模
软件名称:ANSYS2.软件版本:2025R13.软件大小:52.2GB4.安装环境:win10/win11(64位)下载通道:夸克网盘链接:https://pan.quark.cn/s/ce34e3269bd4更多免费软件,游戏等点这里软件介绍ANSYS是一款由ANSYS,Inc.开发的工程仿真软件,广泛应用于结构、流体、电磁、热分析和多物理场耦合等领域。它支持有限元分析(FEA)、计算流体力学
- 【免费下载】 Labview激活软件使用指南
翁丛咏
Labview激活软件使用指南【下载地址】Labview激活软件使用指南本仓库提供了一个Labview激活软件,帮助用户激活Labview软件,使其从试用版变为正式版。Labview是一款强大的图形化编程环境,广泛应用于工程、科研等领域项目地址:https://gitcode.com/open-source-toolkit/b97a8简介本仓库提供了一个Labview激活软件,帮助用户激活Labv
- 有声读书主播是怎么赚钱的?有声读书主播的赚钱之道
高省APP大九
随着有声书市场的蓬勃发展,有声读书主播这一职业逐渐走进大众的视野,成为许多人实现灵活就业和增收的新途径。那么,有声读书主播是如何赚钱的呢?本文将详细探讨这一问题。【高省】APP(高佣金领导者)是一个自用省钱佣金高,分享推广赚钱多的平台,2000万用户信赖的四年老平台,稳定可靠。高省APP佣金更高,模式更好,终端用户不流失。高省是公认的返利最高的软件。蓓蓓导师高省邀请码110000,注册送2皇冠会员
- 工作之余也能赚大钱!上班族必知的2024年副业前五强
高省爱氧惠
当下兼职副业已成为了一个热门词汇,越来越多的朋友都试图通过一份可靠的兼职工作来提高自己的生活质量,同时还能够增加自己的工作经验、促进个人的事业发展,确实是一件一举两得的事情。需求多了,供应自然也就大了,目前市面上的兼职副业数不胜数,其中优劣并存。本期文章就来和大家推荐五个比较不错的兼职副业,适合广大普通人进行值得尝试。1:app拉新App拉新是一个低门槛、高回报的兼职工作,随着市面上app软件的大
- 如何在 Stimulsoft JavaScript 报表组件中,设置设计器与查看器主题风格
CodeCraft Studio
控件报表图表开发javascript开发语言ecmascriptStimulsoftDashboardReport报表仪表盘工具
在现代软件开发中,图形用户界面(GUI)不仅仅是功能的承载体,更是用户体验的关键组成部分。一个美观、统一且具备高度可定制性的界面,能够显著提升系统的专业感和使用效率。Stimulsoft作为功能强大的报表和仪表板解决方案提供商,其JavaScript版本(StimulsoftReports.JS与StimulsoftDashboards.JS)为开发者提供了丰富的内置主题支持,助力快速构建符合品牌
- Text Control 控件教程:使用 .NET C# 中的二维码和条形码增强文档
慧都小妮子
.netc#服务器TXTextControl
QR码和条形码非常适合为文档和PDF文件增加价值,因为它们提供轻松的信息访问、验证信息、跟踪项目和提高交互性。条形码可以弥补纸质或数字人类可读文档与网络门户或网络应用程序中的数字信息之间的差距。大多数用户都熟悉QR码和条形码,它们在许多过程中无处不在,例如:产品包装发票库存管理活动票务登机证支付系统在某些行业中,如果没有条形码,流程将无法进行。这包括医疗保健,可以通过扫描患者佩戴的腕带直接访问患者
- 接口测试流程
鱼鱼说测试
postman
大体流程:3天精通Postman接口测试,全套项目实战教程!!1、(阅读)测试接口文档检验接口文档的完整性、正确性、一致性、易理解性和易浏览性。这个一般在实际测试过程中,都会弱化测试,不注重。2、编写测试用例这个大家都熟,根据接口文档编写测试用例。用例编写方法可以按照黑盒测试的用例编写规则来编写,如:边界值、正交表等等设计方法。3、根据测试用例进行API的手工执行测试根据用例执行测试,注意验证预期
- 如何用 Mockito 玩转单元测试
en-route
单元测试
介绍Mockito是一个广泛使用的Java测试框架,它提供了简洁而强大的功能,用于模拟(mock)和验证对象的行为,尤其是在单元测试中。当我们需要测试某个类的功能时,但又不希望依赖其外部组件或复杂的对象时,可以使用Mockito来创建模拟对象,这些模拟对象可以控制方法返回值、抛出异常或执行特定的逻辑。Mockito使得测试变得更加独立、可靠和可维护,特别是在测试依赖较多或外部系统交互的代码时。从一
- 深入理解设计模式:策略模式的艺术与实践
vvilkin的学习备忘
设计模式设计模式策略模式
在软件开发中,我们经常会遇到需要根据不同情况选择不同算法或行为的场景。传统的做法可能是使用大量的条件语句(if-else或switch-case),但随着需求的增加和变化,这种硬编码的方式会导致代码难以维护和扩展。策略模式(StrategyPattern)正是为了解决这类问题而诞生的一种优雅的设计模式。策略模式属于行为型设计模式,它定义了一系列算法,并将每个算法封装起来,使它们可以相互替换。这种模
- 函数调用栈回溯机制详解
硬核科技
嵌入式单片机开发实战嵌入式嵌入式硬件软件单片机
函数调用回溯Backtrace是现代软件系统调试中的关键技术之一,尤其在嵌入式开发和Linux平台调试中更显重要。它提供了程序在运行或崩溃时的函数调用路径,有助于快速定位错误源。一、函数调用栈与Backtrace的理论基础1.1什么是函数调用栈?函数调用栈(CallStack)是一种由编译器和运行时系统共同维护的后进先出(LIFO)数据结构。每次函数调用时,当前函数的返回地址、局部变量、保存的寄存
- 华为服务器磁盘IO性能查看,磁盘io性能
忘記痛苦
华为服务器磁盘IO性能查看
磁盘io性能内容精选换一换对于不同业务场景,通过在调整数据库的参数配置,可以有效提升服务器性能。使用如下配置文件参数启动数据库,默认配置文件路径为/etc/my.cnf,其中MariaDB软件安装以及数据存放路径根据实际情况修改。根据是否支持挂载至多台云服务器可以将磁盘分为非共享磁盘和共享磁盘。一个非共享磁盘只能挂载至一台云服务器,而一个共享磁盘可以同时挂载至多台云服务器。共享磁盘是一种支持多个云
- 嵌入式开发
王明列
zynqfpga开发
逻辑开发与软件开发,皆为高度专业化的技术领域,能在两者之间自由穿梭、解决复杂问题的工程师,凤毛麟角。然而,“精通”本身并无边界。在实际工程中,无论是算法实现、高速接口,还是雷达系统、电机控制,每一个方向都深邃如海,足以让人终身钻研。真正重要的,从来不是“掌握一切”,而是在关键问题域中,构建起可闭环的解决路径,持续迭代,稳步积累。因为:再庞大的系统,也由一个个“可掌握的知识点”组成;再高的门槛,也能
- 【彻底干净无痕深度卸载Adobe全家桶】Windows11下清理流氓Adobe全家桶
有时候一些特殊原因我们需要卸载干净Adobe,但是幽默Adobe偷偷留下很多答辩藏着不给无脑删除,这时候在Windows11上彻底卸载Adobe系列软件需要结合官方工具和手动清理,以后谁再说360和2345流氓我跟谁急,Adobe比它们流氓一万倍!以下是详细步骤:一、使用Adobe官方卸载工具Adobe提供了专门的清理工具CreativeCloudCleanerTool,可强制移除残留文件和注册表
- LabVIEW VI Server导出功能
LabVIEW中借助VIServer实现导出VI的配置、执行与交互流程,覆盖服务端配置(含权限管理、设置还原)、客户端调用等核心环节,验证跨VI交互与远程调用逻辑,为分布式应用提供基础实现方案。功能说明1.服务端配置(supportExportingVIs-Server.vi关联逻辑)功能:开放本地VIServer服务(基于TCP/IP端口3363),将正弦波(Helper-Export-Sine
- 软件测试面试大全(含答案+文档)
sszmvb1234
面试软件测试面试题软件测试面试职场和发展
Part11、你的测试职业发展是什么?测试经验越多,测试能力越高。所以我的职业发展是需要时间积累的,一步步向着高级测试工程师奔去。而且我也有初步的职业规划,前3年积累测试经验,按如何做好测试工程师的要点去要求自己,不断更新自己改正自己,做好测试任务。优势在于我对测试坚定不移的信心和热情,虽然经验还不够,但测试需要的基本技能我有信心在工作中得以发挥。2、你认为测试人员需要具备哪些素质做测试应该要有一
- Unity UI架构的道与术:从“一团乱麻”到“井然有序”(7)
第六章:架构的回归——在理论的优雅与现实的代价之间,寻找你的最优解穿越了UI技术演进的漫漫长路,我们从一个新手的“一团乱麻”,到用MVC、MVP、MVVM这些“手术刀”,一步步地为代码建立秩序。然而,在这场对“终极优雅”的漫长求索中,我们必须在旅程的终点,停下脚步,回归到所有软件工程最朴素的本质——权衡。架构设计的真谛,不在于找到一个完美的“黄金标准”,而在于清醒地认识到每一种选择背后的代价,并为
- VMware Workstation 11 或者 VMware Player 7安装MAC OS X 10.10 Yosemite
iwindyforest
vmwaremac os10.10workstationplayer
最近尝试了下VMware下安装MacOS 系统,
安装过程中发现网上可供参考的文章都是VMware Workstation 10以下, MacOS X 10.9以下的文章,
只能提供大概的思路, 但是实际安装起来由于版本问题, 走了不少弯路, 所以我尝试写以下总结, 希望能给有兴趣安装OSX的人提供一点帮助。
写在前面的话:
其实安装好后发现, 由于我的th
- 关于《基于模型驱动的B/S在线开发平台》源代码开源的疑虑?
deathwknight
JavaScriptjava框架
本人从学习Java开发到现在已有10年整,从一个要自学 java买成javascript的小菜鸟,成长为只会java和javascript语言的老菜鸟(个人邮箱:
[email protected])
一路走来,跌跌撞撞。用自己的三年多业余时间,瞎搞一个小东西(基于模型驱动的B/S在线开发平台,非MVC框架、非代码生成)。希望与大家一起分享,同时有许些疑虑,希望有人可以交流下
平台
- 如何把maven项目转成web项目
Kai_Ge
mavenMyEclipse
创建Web工程,使用eclipse ee创建maven web工程 1.右键项目,选择Project Facets,点击Convert to faceted from 2.更改Dynamic Web Module的Version为2.5.(3.0为Java7的,Tomcat6不支持). 如果提示错误,可能需要在Java Compiler设置Compiler compl
- 主管???
Array_06
工作
转载:http://www.blogjava.net/fastzch/archive/2010/11/25/339054.html
很久以前跟同事参加的培训,同事整理得很详细,必须得转!
前段时间,公司有组织中高阶主管及其培养干部进行了为期三天的管理训练培训。三天的课程下来,虽然内容较多,因对老师三天来的课程内容深有感触,故借着整理学习心得的机会,将三天来的培训课程做了一个
- python内置函数大全
2002wmj
python
最近一直在看python的document,打算在基础方面重点看一下python的keyword、Build-in Function、Build-in Constants、Build-in Types、Build-in Exception这四个方面,其实在看的时候发现整个《The Python Standard Library》章节都是很不错的,其中描述了很多不错的主题。先把Build-in Fu
- JSP页面通过JQUERY合并行
357029540
JavaScriptjquery
在写程序的过程中我们难免会遇到在页面上合并单元行的情况,如图所示
如果对于会的同学可能很简单,但是对没有思路的同学来说还是比较麻烦的,提供一下用JQUERY实现的参考代码
function mergeCell(){
var trs = $("#table tr");
&nb
- Java基础
冰天百华
java基础
学习函数式编程
package base;
import java.text.DecimalFormat;
public class Main {
public static void main(String[] args) {
// Integer a = 4;
// Double aa = (double)a / 100000;
// Decimal
- unix时间戳相互转换
adminjun
转换unix时间戳
如何在不同编程语言中获取现在的Unix时间戳(Unix timestamp)? Java time JavaScript Math.round(new Date().getTime()/1000)
getTime()返回数值的单位是毫秒 Microsoft .NET / C# epoch = (DateTime.Now.ToUniversalTime().Ticks - 62135
- 作为一个合格程序员该做的事
aijuans
程序员
作为一个合格程序员每天该做的事 1、总结自己一天任务的完成情况 最好的方式是写工作日志,把自己今天完成了什么事情,遇见了什么问题都记录下来,日后翻看好处多多
2、考虑自己明天应该做的主要工作 把明天要做的事情列出来,并按照优先级排列,第二天应该把自己效率最高的时间分配给最重要的工作
3、考虑自己一天工作中失误的地方,并想出避免下一次再犯的方法 出错不要紧,最重
- 由html5视频播放引发的总结
ayaoxinchao
html5视频video
前言
项目中存在视频播放的功能,前期设计是以flash播放器播放视频的。但是现在由于需要兼容苹果的设备,必须采用html5的方式来播放视频。我就出于兴趣对html5播放视频做了简单的了解,不了解不知道,水真是很深。本文所记录的知识一些浅尝辄止的知识,说起来很惭愧。
视频结构
本该直接介绍html5的<video>的,但鉴于本人对视频
- 解决httpclient访问自签名https报javax.net.ssl.SSLHandshakeException: sun.security.validat
bewithme
httpclient
如果你构建了一个https协议的站点,而此站点的安全证书并不是合法的第三方证书颁发机构所签发,那么你用httpclient去访问此站点会报如下错误
javax.net.ssl.SSLHandshakeException: sun.security.validator.ValidatorException: PKIX path bu
- Jedis连接池的入门级使用
bijian1013
redisredis数据库jedis
Jedis连接池操作步骤如下:
a.获取Jedis实例需要从JedisPool中获取;
b.用完Jedis实例需要返还给JedisPool;
c.如果Jedis在使用过程中出错,则也需要还给JedisPool;
packag
- 变与不变
bingyingao
不变变亲情永恒
变与不变
周末骑车转到了五年前租住的小区,曾经最爱吃的西北面馆、江西水饺、手工拉面早已不在,
各种店铺都换了好几茬,这些是变的。
三年前还很流行的一款手机在今天看起来已经落后的不像样子。
三年前还运行的好好的一家公司,今天也已经不复存在。
一座座高楼拔地而起,
- 【Scala十】Scala核心四:集合框架之List
bit1129
scala
Spark的RDD作为一个分布式不可变的数据集合,它提供的转换操作,很多是借鉴于Scala的集合框架提供的一些函数,因此,有必要对Scala的集合进行详细的了解
1. 泛型集合都是协变的,对于List而言,如果B是A的子类,那么List[B]也是List[A]的子类,即可以把List[B]的实例赋值给List[A]变量
2. 给变量赋值(注意val关键字,a,b
- Nested Functions in C
bookjovi
cclosure
Nested Functions 又称closure,属于functional language中的概念,一直以为C中是不支持closure的,现在看来我错了,不过C标准中是不支持的,而GCC支持。
既然GCC支持了closure,那么 lexical scoping自然也支持了,同时在C中label也是可以在nested functions中自由跳转的
- Java-Collections Framework学习与总结-WeakHashMap
BrokenDreams
Collections
总结这个类之前,首先看一下Java引用的相关知识。Java的引用分为四种:强引用、软引用、弱引用和虚引用。
强引用:就是常见的代码中的引用,如Object o = new Object();存在强引用的对象不会被垃圾收集
- 读《研磨设计模式》-代码笔记-解释器模式-Interpret
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
package design.pattern;
/*
* 解释器(Interpreter)模式的意图是可以按照自己定义的组合规则集合来组合可执行对象
*
* 代码示例实现XML里面1.读取单个元素的值 2.读取单个属性的值
* 多
- After Effects操作&快捷键
cherishLC
After Effects
1、快捷键官方文档
中文版:https://helpx.adobe.com/cn/after-effects/using/keyboard-shortcuts-reference.html
英文版:https://helpx.adobe.com/after-effects/using/keyboard-shortcuts-reference.html
2、常用快捷键
- Maven 常用命令
crabdave
maven
Maven 常用命令
mvn archetype:generate
mvn install
mvn clean
mvn clean complie
mvn clean test
mvn clean install
mvn clean package
mvn test
mvn package
mvn site
mvn dependency:res
- shell bad substitution
daizj
shell脚本
#!/bin/sh
/data/script/common/run_cmd.exp 192.168.13.168 "impala-shell -islave4 -q 'insert OVERWRITE table imeis.${tableName} select ${selectFields}, ds, fnv_hash(concat(cast(ds as string), im
- Java SE 第二讲(原生数据类型 Primitive Data Type)
dcj3sjt126com
java
Java SE 第二讲:
1. Windows: notepad, editplus, ultraedit, gvim
Linux: vi, vim, gedit
2. Java 中的数据类型分为两大类:
1)原生数据类型 (Primitive Data Type)
2)引用类型(对象类型) (R
- CGridView中实现批量删除
dcj3sjt126com
PHPyii
1,CGridView中的columns添加
array(
'selectableRows' => 2,
'footer' => '<button type="button" onclick="GetCheckbox();" style=&
- Java中泛型的各种使用
dyy_gusi
java泛型
Java中的泛型的使用:1.普通的泛型使用
在使用类的时候后面的<>中的类型就是我们确定的类型。
public class MyClass1<T> {//此处定义的泛型是T
private T var;
public T getVar() {
return var;
}
public void setVa
- Web开发技术十年发展历程
gcq511120594
Web浏览器数据挖掘
回顾web开发技术这十年发展历程:
Ajax
03年的时候我上六年级,那时候网吧刚在小县城的角落萌生。传奇,大话西游第一代网游一时风靡。我抱着试一试的心态给了网吧老板两块钱想申请个号玩玩,然后接下来的一个小时我一直在,注,册,账,号。
彼时网吧用的512k的带宽,注册的时候,填了一堆信息,提交,页面跳转,嘣,”您填写的信息有误,请重填”。然后跳转回注册页面,以此循环。我现在时常想,如果当时a
- openSession()与getCurrentSession()区别:
hetongfei
javaDAOHibernate
来自 http://blog.csdn.net/dy511/article/details/6166134
1.getCurrentSession创建的session会和绑定到当前线程,而openSession不会。
2. getCurrentSession创建的线程会在事务回滚或事物提交后自动关闭,而openSession必须手动关闭。
这里getCurrentSession本地事务(本地
- 第一章 安装Nginx+Lua开发环境
jinnianshilongnian
nginxluaopenresty
首先我们选择使用OpenResty,其是由Nginx核心加很多第三方模块组成,其最大的亮点是默认集成了Lua开发环境,使得Nginx可以作为一个Web Server使用。借助于Nginx的事件驱动模型和非阻塞IO,可以实现高性能的Web应用程序。而且OpenResty提供了大量组件如Mysql、Redis、Memcached等等,使在Nginx上开发Web应用更方便更简单。目前在京东如实时价格、秒
- HSQLDB In-Process方式访问内存数据库
liyonghui160com
HSQLDB一大特色就是能够在内存中建立数据库,当然它也能将这些内存数据库保存到文件中以便实现真正的持久化。
先睹为快!
下面是一个In-Process方式访问内存数据库的代码示例:
下面代码需要引入hsqldb.jar包 (hsqldb-2.2.8)
import java.s
- Java线程的5个使用技巧
pda158
java数据结构
Java线程有哪些不太为人所知的技巧与用法? 萝卜白菜各有所爱。像我就喜欢Java。学无止境,这也是我喜欢它的一个原因。日常
工作中你所用到的工具,通常都有些你从来没有了解过的东西,比方说某个方法或者是一些有趣的用法。比如说线程。没错,就是线程。或者确切说是Thread这个类。当我们在构建高可扩展性系统的时候,通常会面临各种各样的并发编程的问题,不过我们现在所要讲的可能会略有不同。
- 开发资源大整合:编程语言篇——JavaScript(1)
shoothao
JavaScript
概述:本系列的资源整合来自于github中各个领域的大牛,来收藏你感兴趣的东西吧。
程序包管理器
管理javascript库并提供对这些库的快速使用与打包的服务。
Bower - 用于web的程序包管理。
component - 用于客户端的程序包管理,构建更好的web应用程序。
spm - 全新的静态的文件包管
- 避免使用终结函数
vahoa.ma
javajvmC++
终结函数(finalizer)通常是不可预测的,常常也是很危险的,一般情况下不是必要的。使用终结函数会导致不稳定的行为、更差的性能,以及带来移植性问题。不要把终结函数当做C++中的析构函数(destructors)的对应物。
我自己总结了一下这一条的综合性结论是这样的:
1)在涉及使用资源,使用完毕后要释放资源的情形下,首先要用一个显示的方