STAMP软件安装
软件官网 http://kiwi.cs.dal.ca/Software/STAMP
1. Microsoft Windows系统安装比较简单
2. ubuntu安装
```
# 首先使用apt安装pyqt4
sudo apt install python-qt4
# virtualenv 创建单独的python环境并能访问系统python包(为了使用pyqt4)
virtualenv stamp --python=python2.7 --system-site-packages
# 激活环境
source activate stamp/bin/activate
pip install numpy matplotlib biom-format STAMP
# 使用
./stamp/bin/STAMP
```
也可以使用gnome-desktop-item-edit命令创建快捷方式。下图是stamp环境的python包列表。

conda 安装
```
conda create -n stamp stamp fontconfig freetype icu=56
```
准备数据
16S或者ITS测序分析,最后会得到feature-table(OTU table) 和feature(OTU)的物种注释信息,feature-table通常是biom的格式, 物种注释信息是tab分隔的文本文件。
使用biom软件将两者结合,并转换为tab分隔的文本文件,代码如下:
```
biom add-metadata -i feature-table.biom -o feature-table-tax.biom --observation-metadata-fp taxonomy.tsv --sc-separated taxonomy --observation-header OTUID,taxonomy
biom convert -i feature-table-tax.biom -o feature-table-tax.tsv --to-tsv --table-type "OTU table" --header-key taxonomy
```
结果如下图所示:
因为STAMP需要严格的层级关系,而我们得到的物种注释信息一般满足不了该要求,直接导入STAMP会报错,所以将数据转换为8个级别,分别是OTU,Kingdom,Phylum,Class,Order,Family,Genus,Species,然后在每一个级别上单独分析。在这里使用R进行转换,代码如下:
```
library(tidyverse)
# 导入数据
levels <- c("Kingdom","Phylum","Class","Order","Family","Genus","Species")
data <- read_tsv("feature-table-tax.tsv", skip = 1) %>%
rename(OTU=`#OTU ID`) %>%
mutate(taxonomy = str_replace_all(taxonomy, "[kpcofgs]__", "")) %>%
separate(taxonomy, sep="; ", levels)
# 将没有分类的级别标注为 "unclassified"
data[is.na(data)]="unclassified"
data[data==""]="unclassified"
#Samples number
number <- dim(data)[[2]]-8
#OTU level
otu <- data %>% select(1:(number+1))
write_tsv(otu, "otu.spf")
# Phylum -> Species level
for (level in levels){
n <- which(colnames(data)==level)
temp <- data %>% select(n, 2:(number+1))
colnames(temp)[1]='index'
temp <- temp %>% gather("Sample", "Count", -index) %>%
group_by(index, Sample) %>%
summarise(Count = sum(Count)) %>%
spread(key = Sample, value = Count)
colnames(temp)[1]=level
write_tsv(temp, paste0(level, ".spf", collapse = ""))
}
```
这会生成8个spf文件,其本质是tab分隔的文本文件,这就是要向STAMP导入的数据。
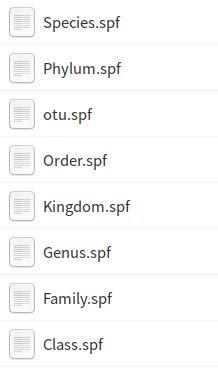
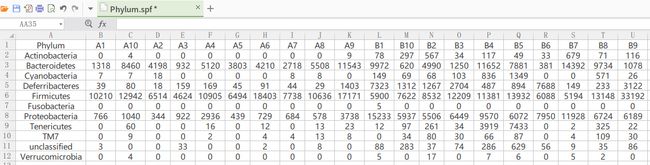
另外还要向STAMP导入实验设计文件,文件内容如下(可以有多列分组信息,例如group, subgroup,这里只有一列):
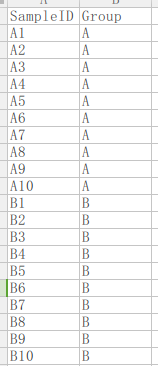
分析
1. 导入数据
从File -> Load data导入数据,以 Genus Level 为例,如下图所示
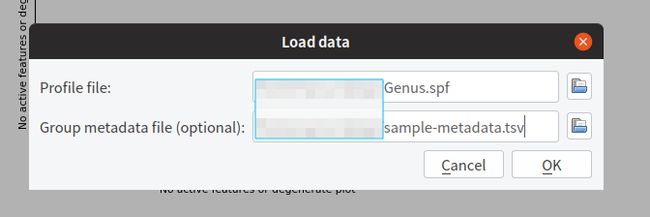
2. 导入数据后可以直接查看PCA分析结果,也可以选择不同的统计分析方法进行分析,支持多组间(>=3组)分析,也可以选择两组或者两个样本比较。

分析结果
统计分析可视化结果和表格均可以导出。


