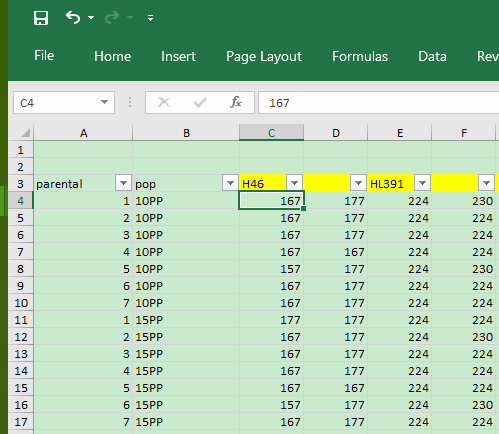
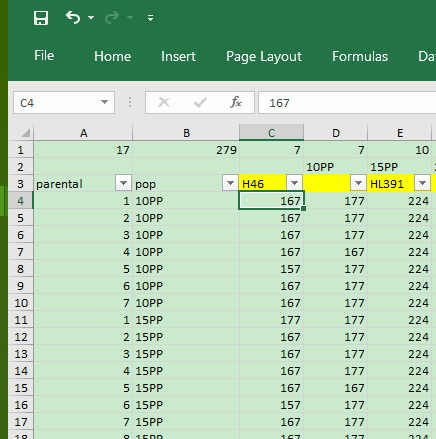
- Linux 系统中常用的文件和文件夹管理命令 and 常用快捷键
高山莫衣
gitlinux运维服务器
以下是Linux系统中常用的文件和文件夹管理命令,分类整理便于快速查阅:目录操作命令作用示例pwd显示当前工作目录pwdcd切换目录cd/var/wwwmkdir创建目录mkdirnew_foldermkdir-p递归创建多级目录mkdir-pa/b/crmdir删除空目录rmdirempty_dirtree树状显示目录结构tree-L2(显示2层深度)文件操作命令作用示例ls列出目录内容ls-l
- 思维树(Tree of Thoughts): 超越链式思维的AI推理新范式
司南锤
LLM人工智能
引言在人工智能快速发展的今天,大语言模型(LLM)的推理能力一直是研究的热点。从最初的直接问答,到链式思维(ChainofThoughts,CoT)的出现,再到如今的思维树(TreeofThoughts,TOT),AI的推理方式正在变得越来越接近人类的思维过程。思维树作为一种全新的推理框架,不仅继承了链式思维的优势,更通过树状结构的探索和回溯机制,实现了更加复杂和深入的推理过程。本文将深入探讨TO
- Swift 实现二叉树垂直遍历:LeetCode 314 完整解析与实战示例
网罗开发
Swiftswiftleetcode开发语言
文章目录摘要描述题解答案题解代码分析代码关键点解释:示例测试及结果解释一下输出:时间复杂度空间复杂度总结摘要在日常项目中,处理「树状结构」并不是稀罕事,比如做组织架构图、文件夹视图或者评论嵌套列表,我们经常会遇到对树的各种“奇怪”遍历方式。今天这题LeetCode314——BinaryTreeVerticalOrderTraversal(二叉树的垂直遍历),就考验了我们如何按垂直方向组织二叉树节点
- 1143 Lowest Common Ancestor (30 分)
Thelowestcommonancestor(LCA)oftwonodesUandVinatreeisthedeepestnodethathasbothUandVasdescendants.Abinarysearchtree(BST)isrecursivelydefinedasabinarytreewhichhasthefollowingproperties:Theleftsubtreeofan
- 1143 Lowest Common Ancestor (30 分)
依久_
PAT甲
Thelowestcommonancestor(LCA)oftwonodesUandVinatreeisthedeepestnodethathasbothUandVasdescendants.Abinarysearchtree(BST)isrecursivelydefinedasabinarytreewhichhasthefollowingproperties:Theleftsubtreeofan
- 1151 LCA in a Binary Tree (30)
Thelowestcommonancestor(LCA)oftwonodesUandVinatreeisthedeepestnodethathasbothUandVasdescendants.Givenanytwonodesinabinarytree,youaresupposedtofindtheirLCA.InputSpecification:Eachinputfilecontainsonete
- Java基础 集合框架 之Set框架之TreeSet
骑牛小道士
集合框架之Setjava开发语言
TreeSetTreeSet数据结构及实现原理TreeSet的构造方法TreeSet核心特性有序性(`排序大小输出`)自然排序定制排序唯一性底层数据结构:红黑树导航方法(特色核心优势)基础导航方法范围视图(不修改原集合)提取和删除元素逆序视图不允许null元素TreeSet线程不安全TreeSet线程不安全体现解决方案TreeSet优缺点TreeSet应用场景类结构传承去区别于HashSet实现了
- 用Rust编写的开源支付解决方案——Hyperswitch
Hyperswitch是一家全球支付转换公司,旨在简化和优化企业的支付操作。它提供了一个统一的平台来管理各种支付处理器之间的交易,包括Adyen、Braintree、PayPal、Worldpay、Fiserv、Stripe、Authorize.net和Checkout。Stars数21,111Forks数3,514主要特点单一API集成:通过统一的API连接多个支付处理器,无需进行多次集成操作智
- LeetCode高频100题刷题记录之——二叉树的中序遍历
巍巍微澜
Leetcode刷题记录leetcode算法python二叉树
1问题描述给定一个二叉树,按照左,中,右的顺序遍历这棵树。2代码实现思路很简单,从左到右遍历这颗二叉树即可。2.1递归代码实现#Definitionforabinarytreenode.#classTreeNode:#def__init__(self,val=0,left=None,right=None):#self.val=val#self.left=left#self.right=right#
- 二叉树题解——二叉树的中序遍历【LeetCode】统一写法版本
94.二叉树的中序遍历一、算法逻辑(逐步通顺地讲解)这段代码的目标是实现中序遍历,即按照顺序:左子树→当前节点→右子树遍历整个二叉树,并返回节点值的列表。与常见的递归或传统栈方法不同,这里使用的是一种“统一写法”技巧,将“节点值访问”与“节点展开”分开处理,流程如下:1️⃣初始化结构使用一个栈保存待处理元素(可能是TreeNode或int);初始栈中放入整棵树的根节点;结果数组rst用来保存最终遍
- 树结构和数组之间的转化
weixin_45907435
javascript开发语言ecmascript
1、树结构转为数组treeToArray(treeData,returnValue=[]){letnewValue=[...returnValue]treeData.map(item=>{if(item.children){const{children,...treeObj}={...item}newValue.push(treeObj)newValue=this.treeToArray(chil
- Linux基础命令集合
牛岚风
linux运维服务器
目录文件目录相关命令lscdcpfindmkdirmvrmtouchfiletreechattrlsattrmd5sum查看文件以及内容处理相关命令vimcatmore和headtailcutsortuniqwcgreptr文件压缩以及解压缩相关命令tarunzipgzipzip软件包管理相关命令rpmyumapt-get信息显示相关命令unamehostnameuptimestatdudftop
- 144. 二叉树的前序遍历 145. 二叉树的后序遍历 94. 二叉树的中序遍历(多种解法的进阶)
小可爱amour
每日一题算法技巧leetcode二叉树
144.二叉树的前序遍历题目:给定一个二叉树,返回它的前序遍历。示例:输入:[1,null,2,3]1\2/3输出:[1,2,3]解法1:递归classSolution{private:voidtakeVal(TreeNode*root,vector&res){if(NULL==root)return;res.push_back(root->val);takeVal(root->left,res)
- GO(1):GoLand GOPATH和GOROOT详解
rockywish
gogolang
本文所涉及代码路径:https://gitee.com/rockywish/go/tree/master/gopath一、GOPATH的作用第一方:当前工程,第二方:SDK,除此以外的就是第三方存放SDK以外的第三方类库、可以是下载的第三方类库也可以是自己收藏的可复用代码二、配置路径:window:File->Setting->Go->GOPATHmac:Preferences->Go->GOPA
- MySQL的btree索引和hash索引的区别
xiaolyuh123
MySQL哈希算法mysql算法
MySQL的BTree索引和Hash索引的区别一、定义类型定义说明时间复杂度BTree索引使用B+树结构组织索引数据,适用于范围查询、有序遍历等O(logn)Hash索引使用哈希表结构组织索引,仅适用于等值查找操作O(1)二、使用引擎存储引擎索引类型InnoDB默认使用BTree索引Memory默认使用Hash索引,可手动改为BTree三、核心区别对比维度BTree索引Hash索引数据结构B+树结
- P1967 [NOIP 2013 提高组] 货车运输(树链剖分+线段树)
gw_water
cocoac++算法贪心算法数据结构
文章目录题目要求一、解题思路二、解题过程1.数据结构2.求最小生成树(Kruskal算法)2.答案计算(TCD+SegementTree)AC代码题目要求A国有n座城市,编号从1到n,城市之间有m条双向道路。每一条道路对车辆都有重量限制,简称限重。现在有q辆货车在运输货物,司机们想知道每辆车在不超过车辆限重的情况下,最多能运多重的货物。一、解题思路本题求一条路径,使得其在不超过限制重量的前提下,载
- js递归树结构,返回符合条件的子集
啃火龙果的兔子
开发DEMOjavascript开发语言ecmascript
JavaScript递归遍历树结构返回符合条件的子集下面我将介绍几种在JavaScript中递归遍历树结构并返回符合条件的子集的方法。方法一:使用递归函数返回符合条件的子树functionfindSubtree(tree,condition){if(condition(tree)){returntree;}if(tree.children&&tree.children.length){for(le
- [257] 二叉树的所有路径
紫菜(Nori)
数据结构与算法细节TODO算法数据结构leetcode
利用树的先序遍历,采用递归和迭代方式实现迭代方式有待优化/**@lcapp=leetcode.cnid=257lang=java**[257]二叉树的所有路径*///@lccode=start/***Definitionforabinarytreenode.*publicclassTreeNode{*intval;*TreeNodeleft;*TreeNoderight;*TreeNode(){}
- InnoDB 索引数据结构的详解
lanbing
Mysql数据结构mysql
InnoDB存储引擎的索引结构基于B+树(B+Tree),这是其核心特性之一。B+树的设计结合了磁盘存储特性和数据库查询需求,能够高效地处理大规模数据的查找、插入、删除和范围查询操作。以下是InnoDB索引数据结构的详细说明:1.B+树的结构特点B+树是一种自平衡的多路搜索树,其核心特性如下:所有数据存储在叶子节点:B+树的非叶子节点仅存储键值(Key)和子节点指针,而实际的数据(记录)只存在于叶
- 每天一个前端小知识 Day 16 - 前端性能优化全流程指南
蓝婷儿
前端面试前端性能优化
前端性能优化全流程指南(从加载到交互)目标概览:前端性能优化四大核心维度阶段优化目标加载阶段首屏速度、资源压缩、请求优化渲染阶段减少回流重绘、避免布局抖动交互阶段保持高帧率、避免卡顿持久运行阶段内存泄露处理、缓存命中策略一、加载性能优化(首屏速度为王)✅核心策略:资源体积优化JS/CSS/图片压缩(如gzip,brotli)Tree-shaking(去除无用代码)图片压缩(webp优先)合理拆包(
- 数据结构学习——树的储存结构
uwvwko
数据库学习算法树
三种表示法:双亲表示法,孩子表示法,孩子兄弟表示法双亲表示法//树结构——双亲表示法#includeusingnamespacestd;structTree{stringdata;Tree*parent;//双亲指针Tree*firstchild;//第一个孩子指针Tree*nextsibling;//下一个兄弟指针};voidCreateTree(Tree*&root,stringdata,Tr
- Git常见使用
北珣.
git
基本操作创建仓库1.先创建一个文件,再进入到对应的文件夹中#创建文件mkdir[file_name]#进入该文件cd[file_name]2.创建对应的Git仓库(在对应的文件夹内)#创建对应的仓库gitinit#可以查看当前文件内的内容llfile_name#查看tree目录tree.git/配置本地仓库必须要配置的配置项:nameemail为了方便操作,推荐在初始化仓库之后就进行配置#配置gi
- 记录一个异常检测库
STO检测王
深度学习
https://github.com/openvinotoolkit/anomalib/tree/main关于一个异常检测库,包括最先进的算法和功能,如实验管理,超参数优化和边缘推理。
- 家谱html源码,好看的族谱树状图效果代码
Illusion.H
家谱html源码
家谱树状代码demobywww.webym.net/*NowtheCSS*/*{margin:0;padding:0;}.treeul{padding-top:20px;position:relative;transition:all0.5s;-webkit-transition:all0.5s;-moz-transition:all0.5s;}.treeli{float:left;text-al
- 红黑树与2-3树:插入、删除操作的时间复杂度与实现机制比较
一键难忘
红黑树数据结构
本文收录于专栏:算法之翼红黑树与2-3树:插入、删除操作的时间复杂度与实现机制比较红黑树(Red-BlackTree)和2-3树(2-3Tree)是两种广泛用于平衡二叉查找树的自平衡树结构。它们在插入、删除和查找操作中的性能都表现良好,并且可以确保树的高度是对数级别,从而保证了高效的操作时间。本文将对红黑树和2-3树进行深入的比较,并结合代码实例说明它们的实现和应用。1.数据结构简介1.1红黑树简
- Python网安-zip文件暴力破解(仅供学习)
Whoisshutiao
python网安python开发语言网络安全
目录源码在这里需要的模块准备一个密码本和需要破解的ZIP文件一行一行地从密码文件中读取每个密码。核心部分注意,需要修改上段代码注释里的这段具有编码问题的代码:源码在这里https://github.com/Wist-fully/Attack/tree/cracker需要的模块fromtqdmimporttqdmimportzipfileimportpyzipper准备一个密码本和需要破解的ZIP文
- Python网安-ftp服务暴力破解(仅供学习)
Whoisshutiao
python网络安全开发语言
目录源码在这里需要导入的模块连接ftp,并设置密码本和线程核心代码设置线程源码在这里https://github.com/Wist-fully/Attack/tree/cracker需要导入的模块importftplibfromthreadingimportThreadimportqueue连接ftp,并设置密码本和线程host="192.168.6.6"user="student"port=21
- memory_info:Flutter 插件助力鸿蒙生态,精准获取设备内存信息
harmonyos
memory_info:Flutter插件助力鸿蒙生态,精准获取设备内存信息帮助您获取设备内存信息(ram&rom)本项目作者:王阳科/坚果您可以使用这个Flutter插件来更改应用程序图标上的角标作者仓库:https://github.com/MrOlolo/memory_info/tree/master/memory_info在数字化浪潮的推动下,跨平台开发框架如Flutter凭借其高效、便捷
- Leetcode 3600. Maximize Spanning Tree Stability with Upgrades
Espresso Macchiato
leetcode笔记leetcode3600leetcodehardleetcode周赛456二分法DSUUF并查集
Leetcode3600.MaximizeSpanningTreeStabilitywithUpgrades1.解题思路2.代码实现题目链接:3600.MaximizeSpanningTreeStabilitywithUpgrades1.解题思路这一题核心思路就是一个二分法的思路。我们定义函数is_possible(x),表示是否存在一个树的构造,使得任意一条边的长度均不少于xxx。显然,这里有两
- 力扣打卡第十五天 层次遍历非递归+递归
??tobenewyorker
算法leetcode职场和发展
102.二叉树的层序遍历给你二叉树的根节点root,返回其节点值的层序遍历。(即逐层地,从左到右访问所有节点)。示例1:输入:root=[3,9,20,null,null,15,7]输出:[[3],[9,20],[15,7]]示例2:输入:root=[1]输出:[[1]]示例3:输入:root=[]输出:[]提示:树中节点数目在范围[0,2000]内-1000>levelOrder(TreeNod
- PHP如何实现二维数组排序?
IT独行者
二维数组PHP排序
二维数组在PHP开发中经常遇到,但是他的排序就不如一维数组那样用内置函数来的方便了,(一维数组排序可以参考本站另一篇文章【PHP中数组排序函数详解汇总】)。二维数组的排序需要我们自己写函数处理了,这里UncleToo给大家分享一个PHP二维数组排序的函数:
代码:
functionarray_sort($arr,$keys,$type='asc'){
$keysvalue= $new_arr
- 【Hadoop十七】HDFS HA配置
bit1129
hadoop
基于Zookeeper的HDFS HA配置主要涉及两个文件,core-site和hdfs-site.xml。
测试环境有三台
hadoop.master
hadoop.slave1
hadoop.slave2
hadoop.master包含的组件NameNode, JournalNode, Zookeeper,DFSZKFailoverController
- 由wsdl生成的java vo类不适合做普通java vo
darrenzhu
VOwsdlwebservicerpc
开发java webservice项目时,如果我们通过SOAP协议来输入输出,我们会利用工具从wsdl文件生成webservice的client端类,但是这里面生成的java data model类却不适合做为项目中的普通java vo类来使用,当然有一中情况例外,如果这个自动生成的类里面的properties都是基本数据类型,就没问题,但是如果有集合类,就不行。原因如下:
1)使用了集合如Li
- JAVA海量数据处理之二(BitMap)
周凡杨
java算法bitmapbitset数据
路漫漫其修远兮,吾将上下而求索。想要更快,就要深入挖掘 JAVA 基础的数据结构,从来分析出所编写的 JAVA 代码为什么把内存耗尽,思考有什么办法可以节省内存呢? 啊哈!算法。这里采用了 BitMap 思想。
首先来看一个实验:
指定 VM 参数大小: -Xms256m -Xmx540m
- java类型与数据库类型
g21121
java
很多时候我们用hibernate的时候往往并不是十分关心数据库类型和java类型的对应关心,因为大多数hbm文件是自动生成的,但有些时候诸如:数据库设计、没有生成工具、使用原始JDBC、使用mybatis(ibatIS)等等情况,就会手动的去对应数据库与java的数据类型关心,当然比较简单的数据类型即使配置错了也会很快发现问题,但有些数据类型却并不是十分常见,这就给程序员带来了很多麻烦。
&nb
- Linux命令
510888780
linux命令
系统信息
arch 显示机器的处理器架构(1)
uname -m 显示机器的处理器架构(2)
uname -r 显示正在使用的内核版本
dmidecode -q 显示硬件系统部件 - (SMBIOS / DMI)
hdparm -i /dev/hda 罗列一个磁盘的架构特性
hdparm -tT /dev/sda 在磁盘上执行测试性读取操作
cat /proc/cpuinfo 显示C
- java常用JVM参数
墙头上一根草
javajvm参数
-Xms:初始堆大小,默认为物理内存的1/64(<1GB);默认(MinHeapFreeRatio参数可以调整)空余堆内存小于40%时,JVM就会增大堆直到-Xmx的最大限制
-Xmx:最大堆大小,默认(MaxHeapFreeRatio参数可以调整)空余堆内存大于70%时,JVM会减少堆直到 -Xms的最小限制
-Xmn:新生代的内存空间大小,注意:此处的大小是(eden+ 2
- 我的spring学习笔记9-Spring使用工厂方法实例化Bean的注意点
aijuans
Spring 3
方法一:
<bean id="musicBox" class="onlyfun.caterpillar.factory.MusicBoxFactory"
factory-method="createMusicBoxStatic"></bean>
方法二:
- mysql查询性能优化之二
annan211
UNIONmysql查询优化索引优化
1 union的限制
有时mysql无法将限制条件从外层下推到内层,这使得原本能够限制部分返回结果的条件无法应用到内层
查询的优化上。
如果希望union的各个子句能够根据limit只取部分结果集,或者希望能够先排好序在
合并结果集的话,就需要在union的各个子句中分别使用这些子句。
例如 想将两个子查询结果联合起来,然后再取前20条记录,那么mys
- 数据的备份与恢复
百合不是茶
oraclesql数据恢复数据备份
数据的备份与恢复的方式有: 表,方案 ,数据库;
数据的备份:
导出到的常见命令;
参数 说明
USERID 确定执行导出实用程序的用户名和口令
BUFFER 确定导出数据时所使用的缓冲区大小,其大小用字节表示
FILE 指定导出的二进制文
- 线程组
bijian1013
java多线程threadjava多线程线程组
有些程序包含了相当数量的线程。这时,如果按照线程的功能将他们分成不同的类别将很有用。
线程组可以用来同时对一组线程进行操作。
创建线程组:ThreadGroup g = new ThreadGroup(groupName);
&nbs
- top命令找到占用CPU最高的java线程
bijian1013
javalinuxtop
上次分析系统中占用CPU高的问题,得到一些使用Java自身调试工具的经验,与大家分享。 (1)使用top命令找出占用cpu最高的JAVA进程PID:28174 (2)如下命令找出占用cpu最高的线程
top -Hp 28174 -d 1 -n 1
32694 root 20 0 3249m 2.0g 11m S 2 6.4 3:31.12 java
- 【持久化框架MyBatis3四】MyBatis3一对一关联查询
bit1129
Mybatis3
当两个实体具有1对1的对应关系时,可以使用One-To-One的进行映射关联查询
One-To-One示例数据
以学生表Student和地址信息表为例,每个学生都有都有1个唯一的地址(现实中,这种对应关系是不合适的,因为人和地址是多对一的关系),这里只是演示目的
学生表
CREATE TABLE STUDENTS
(
- C/C++图片或文件的读写
bitcarter
写图片
先看代码:
/*strTmpResult是文件或图片字符串
* filePath文件需要写入的地址或路径
*/
int writeFile(std::string &strTmpResult,std::string &filePath)
{
int i,len = strTmpResult.length();
unsigned cha
- nginx自定义指定加载配置
ronin47
进入 /usr/local/nginx/conf/include 目录,创建 nginx.node.conf 文件,在里面输入如下代码:
upstream nodejs {
server 127.0.0.1:3000;
#server 127.0.0.1:3001;
keepalive 64;
}
server {
liste
- java-71-数值的整数次方.实现函数double Power(double base, int exponent),求base的exponent次方
bylijinnan
double
public class Power {
/**
*Q71-数值的整数次方
*实现函数double Power(double base, int exponent),求base的exponent次方。不需要考虑溢出。
*/
private static boolean InvalidInput=false;
public static void main(
- Android四大组件的理解
Cb123456
android四大组件的理解
分享一下,今天在Android开发文档-开发者指南中看到的:
App components are the essential building blocks of an Android
- [宇宙与计算]涡旋场计算与拓扑分析
comsci
计算
怎么阐述我这个理论呢? 。。。。。。。。。
首先: 宇宙是一个非线性的拓扑结构与涡旋轨道时空的统一体。。。。
我们要在宇宙中寻找到一个适合人类居住的行星,时间非常重要,早一个刻度和晚一个刻度,这颗行星的
- 同一个Tomcat不同Web应用之间共享会话Session
cwqcwqmax9
session
实现两个WEB之间通过session 共享数据
查看tomcat 关于 HTTP Connector 中有个emptySessionPath 其解释如下:
If set to true, all paths for session cookies will be set to /. This can be useful for portlet specification impleme
- springmvc Spring3 MVC,ajax,乱码
dashuaifu
springjquerymvcAjax
springmvc Spring3 MVC @ResponseBody返回,jquery ajax调用中文乱码问题解决
Spring3.0 MVC @ResponseBody 的作用是把返回值直接写到HTTP response body里。具体实现AnnotationMethodHandlerAdapter类handleResponseBody方法,具体实
- 搭建WAMP环境
dcj3sjt126com
wamp
这里先解释一下WAMP是什么意思。W:windows,A:Apache,M:MYSQL,P:PHP。也就是说本文说明的是在windows系统下搭建以apache做服务器、MYSQL为数据库的PHP开发环境。
工欲善其事,必须先利其器。因为笔者的系统是WinXP,所以下文指的系统均为此系统。笔者所使用的Apache版本为apache_2.2.11-
- yii2 使用raw http request
dcj3sjt126com
http
Parses a raw HTTP request using yii\helpers\Json::decode()
To enable parsing for JSON requests you can configure yii\web\Request::$parsers using this class:
'request' =&g
- Quartz-1.8.6 理论部分
eksliang
quartz
转载请出自出处:http://eksliang.iteye.com/blog/2207691 一.概述
基于Quartz-1.8.6进行学习,因为Quartz2.0以后的API发生的非常大的变化,统一采用了build模式进行构建;
什么是quartz?
答:简单的说他是一个开源的java作业调度框架,为在 Java 应用程序中进行作业调度提供了简单却强大的机制。并且还能和Sp
- 什么是POJO?
gupeng_ie
javaPOJO框架Hibernate
POJO--Plain Old Java Objects(简单的java对象)
POJO是一个简单的、正规Java对象,它不包含业务逻辑处理或持久化逻辑等,也不是JavaBean、EntityBean等,不具有任何特殊角色和不继承或不实现任何其它Java框架的类或接口。
POJO对象有时也被称为Data对象,大量应用于表现现实中的对象。如果项目中使用了Hiber
- jQuery网站顶部定时折叠广告
ini
JavaScripthtmljqueryWebcss
效果体验:http://hovertree.com/texiao/jquery/4.htmHTML文件代码:
<!DOCTYPE html>
<html xmlns="http://www.w3.org/1999/xhtml">
<head>
<title>网页顶部定时收起广告jQuery特效 - HoverTree<
- Spring boot内嵌的tomcat启动失败
kane_xie
spring boot
根据这篇guide创建了一个简单的spring boot应用,能运行且成功的访问。但移植到现有项目(基于hbase)中的时候,却报出以下错误:
SEVERE: A child container failed during start
java.util.concurrent.ExecutionException: org.apache.catalina.Lif
- leetcode: sort list
michelle_0916
Algorithmlinked listsort
Sort a linked list in O(n log n) time using constant space complexity.
====analysis=======
mergeSort for singly-linked list
====code======= /**
* Definition for sin
- nginx的安装与配置,中途遇到问题的解决
qifeifei
nginx
我使用的是ubuntu13.04系统,在安装nginx的时候遇到如下几个问题,然后找思路解决的,nginx 的下载与安装
wget http://nginx.org/download/nginx-1.0.11.tar.gz
tar zxvf nginx-1.0.11.tar.gz
./configure
make
make install
安装的时候出现
- 用枚举来处理java自定义异常
tcrct
javaenumexception
在系统开发过程中,总少不免要自己处理一些异常信息,然后将异常信息变成友好的提示返回到客户端的这样一个过程,之前都是new一个自定义的异常,当然这个所谓的自定义异常也是继承RuntimeException的,但这样往往会造成异常信息说明不一致的情况,所以就想到了用枚举来解决的办法。
1,先创建一个接口,里面有两个方法,一个是getCode, 一个是getMessage
public
- erlang supervisor分析
wudixiaotie
erlang
当我们给supervisor指定需要创建的子进程的时候,会指定M,F,A,如果是simple_one_for_one的策略的话,启动子进程的方式是supervisor:start_child(SupName, OtherArgs),这种方式可以根据调用者的需求传不同的参数给需要启动的子进程的方法。和最初的参数合并成一个数组,A ++ OtherArgs。那么这个时候就有个问题了,既然参数不一致,那