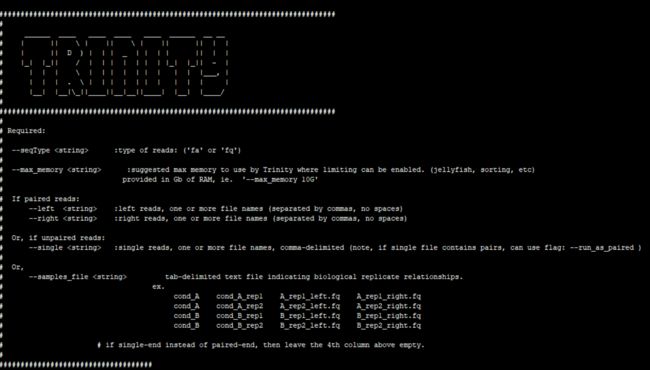
- C++11中的std::ratio:编译时有理数运算的艺术
文章目录一、ratio的核心设计:编译时分数表示1.1自动约分机制1.2符号规范化二、编译时算术运算:ratio的代数体系2.1运算示例2.2编译时验证三、比例比较:编译时逻辑判断四、SI单位体系:预定义比例的实际应用五、实战应用:构建类型安全的单位系统六、注意事项与局限性6.1编译时错误处理6.2与浮点数的对比七、C++26扩展:更小与更大的单位结语在C++11标准中,引入了许多强大的模板元编程
- supervisord:使用RPC管理进程资源实战指南
京脉圈
本文还有配套的精品资源,点击获取简介:supervisord是一个用于管理Unix环境下进程的开源工具,支持进程的启动、停止、重启和异常恢复。本文深入探讨supervisord及其与RPC的结合使用,涉及JAVA、MAVEN和Spring在进程管理中的应用。通过配置文件和XML-RPC接口,实现远程管理进程的便捷性,尤其适用于分布式系统和微服务架构,提高开发者对后台服务的监控和控制效率。1.sup
- 服务治理技术深度解析
我是廖志伟
Java场景面试宝典ServiceGovernanceMicroservicesArchitectureConfigurationManagement
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- ShardingSphere 深度解析
我是廖志伟
Java场景面试宝典ShardingSphereDistributedDatabaseMiddleware
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- JVM与Spring Boot深度解析
我是廖志伟
Java场景面试宝典JavaJVMSpringBoot
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- 并发编程与MyBatis核心解析
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- 使用美团NoCode生成应用-实战案例(一)
零代码探险家
AI编程人工智能数据库react.jsreactjssql
一、概要通过NoCode搭建的值班管理神器,可以支持人员信息批量导入、自定义排班周期,一键生成规则化值班表,适配多角色分工,更可实时导出表格,彻底告别手动排班烦恼!二、实现路径拆解需求输入:明确值班表内容(如日期、人员、岗位)及预设排班规则(如工作日与非工作日所需值班人数)。迭代优化:针对初版人员识别偏差,重构输入逻辑(如多人员批量导入格式优化),通过差异分析精准定位问题。功能完善:新增名单导入模
- JVM与Spring Boot核心解析
Java廖志伟
Java场景面试宝典JavaJVMPerformanceOptimization
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- 互联网大厂Java面试指南:从基础到高阶技术栈与业务场景实战
互联网大厂Java面试指南:从基础到高阶技术栈与业务场景实战第一轮:Java基础与Spring生态问题1:请解释Java中的多线程实现方式及其适用场景。解析:核心概念:Java多线程可通过继承Thread类或实现Runnable接口实现,推荐后者以避免单继承限制。ExecutorService是更高级的线程池管理工具。适用场景:高并发任务如电商秒杀、实时数据处理。底层机制:JVM线程模型基于操作系
- SEO优化技巧深度解析:从算法逻辑到实战策略的全链路突破
boyedu
网站建设网站建设网站运营网站架构
第一章搜索引擎算法逻辑:SEO优化的底层密码1.1算法进化史:从关键词匹配到意图理解搜索引擎算法经历了从简单关键词匹配到复杂语义理解的跨越式发展。早期算法以PageRank为核心,通过分析网页间链接关系评估权威性。随着Hummingbird算法的推出,搜索引擎开始解析自然语言,BERT算法进一步实现上下文语义理解。当前算法已形成多维度评估体系,涵盖内容质量、用户体验、权威性建设等层面。以Googl
- 有了 25k Star 的MediaCrawler爬虫库加持,三分钟搞定某红书、某音等平台爬取!
前端后端爬虫
大家好,我是程序员凌览。今天给大家介绍一个超实用的Python爬虫实战项目——MediaCrawler。这个项目可以实现小红书、抖音、快手、B站和微博的爬虫功能,覆盖了当下热门的自媒体平台。它能够高效抓取这些平台上的视频、图片、评论、点赞和转发等信息。MediaCrawler支持的平台及功能如下图:快速开始下载项目代码访问MediaCrawlerGitHub仓库,点击“Code”按钮下载项目代码。
- 如何道破信息差,精准准备秋招?——应届生秋招全流程解析
信息差,是大多数应届生秋招失败的关键。本文将从认知差距、平台渠道、实战建议三大维度,帮你打破信息壁垒,走上offer收割之路。一、什么是秋招中的“信息差”?在秋招中,应届生之间的差距并非仅仅是技术实力,更在于“知道什么”和“知道怎么做”的能力差异。常见的信息差类型:信息差类型表现形式企业信息差不知道哪些公司在招人,不清楚岗位要求面试流程差不知道面试题类型、不清楚流程节点技术准备差不知道八股文、项目
- 接口漏洞怎么抓?Fiddler 中文版 + Postman + Wireshark 实战指南
2501_91591841
httpudphttpswebsocket网络安全网络协议tcp/ip
接口安全是现代应用开发中的高危环节:一旦API存在未授权访问、参数篡改、权限绕过等漏洞,可能直接导致用户信息泄露、资金损失甚至整个平台瘫痪。对于开发和安全人员来说,光依赖后端日志排查远远不够,需要对接口进行主动安全性验证。而Fiddler抓包工具提供了灵活的请求拦截、修改、重放功能,是在API安全防护与漏洞复现中必不可少的工具。再结合Postman、Wireshark等工具,可以从接口到网络层做全
- SQL查询实战:高效数据检索全攻略
六七_Shmily
数据库sql数据库
SQLDQL详解:SELECT查询与核心子句DQL(DataQueryLanguage)是SQL中最常用的部分,核心是SELECT语句,用于从数据库检索数据。以下是完整结构和各子句的详细解析:SELECT[DISTINCT]column_list--选择字段FROMtable_sources--数据来源[JOIN_TYPEJOINtableONjoin_condition]--多表连接[WHERE
- 基于JAVA+SpringBoot+Vue+Echarts的充电数据大屏可视化分析
✌全网粉丝20W+,csdn特邀作者、博客专家、CSDN新星计划导师、java领域优质创作者,博客之星、掘金/华为云/阿里云/InfoQ等平台优质作者、专注于Java技术领域和毕业项目实战✌文末获取项目下载方式一、项目背景介绍:随着电动汽车的普及,城市中充电设施的需求日益增长。为了提高充电设施的管理效率和用户体验,本文提出了一个停车场充电桩数据可视化平台的设计与实现。该平台旨在集成、处理并展示来自
- 《深入浅出 React 19:AI 视角下的源码解析与进阶》- JSX 与 React Element
如果你对React源码解析感兴趣,欢迎访问我的个人博客:深入浅出React19:AI视角下的源码解析与进阶或者我的微信公众号-前端小卒在我的博客和公众号中,你可以找到:完整的React源码解析电子书-从基础概念到高级实现,全面覆盖React18的核心机制系统化的学习路径-按照React的执行流程,循序渐进地深入每个模块实战案例分析-结合真实场景,理解React设计思想和最佳实践最新技术动态-持续更
- 云原生API Gateway:连接微服务的桥梁
AI云原生与云计算技术学院
云原生gateway微服务ai
云原生APIGateway:连接微服务的桥梁关键词:云原生、API网关、微服务架构、服务治理、流量管理、服务网格、DevOps摘要:本文深入探讨云原生环境下API网关的核心原理与实践应用,解析其在微服务架构中作为统一入口的关键作用。通过详细阐述API网关的核心功能、技术架构、算法原理及数学模型,结合Kubernetes实战案例演示流量管理、安全防护、服务编排等核心能力。同时分析典型应用场景,推荐前
- 【Python】Python类型标注革命:Annotated类型深度解析与实战
田辛 | 田豆芽
Pythonpython设计模式类型驱动设计
一、初识Annotated:类型系统的拓展革命作为深耕Python领域多年的开发者,田辛老师在第一次接触typing.Annotated时的感受可以用"惊艳"来形容。这个Python3.9引入的类型构造器,为我们打开了元数据整合的新维度。基本语法结构:fromtypingimportAnnotatedTemperature=Annotated[float,"Celsius"]这里我们创建了一个带有
- JavaScript高程设计第一章---什么是JavaScript
小顾万家
javascript
文章目录前言一、JavaScript实现二、ECMAScript1.ECMAScript概念2.ECMAScript版本3.ECMAScript符合性三、DOM1.DOM概念2.DOM级别三、BOM1.BOM概念前言通过自身对前端的学习和认知,发现仅仅通过看教学视频来学习前端是不够的,还需要通过阅读相关的前端书籍来扩大自己的知识面。今天我就来总结一下自己通过阅读《JavaScript高级程序设计》
- Android15音频进阶之高通Adsp触发ramdump(一百二十六)
Android系统攻城狮
AndroidAudio工程师进阶系列Android15音频进阶高通平台
简介:CSDN博客专家、《Android系统多媒体进阶实战》一书作者博主新书推荐:《Android系统多媒体进阶实战》AndroidAudio工程师专栏:Audio工程师进阶系列【原创干货持续更新中……】Android多媒体专栏:多媒体系统工程师系列【原创干货持续更新中……】推荐1:车载系统实战课:
- PyTorch深度学习优化实战:从理论到实践的现代化技能指南
智算菩萨
深度学习pytorch人工智能
引言:现代PyTorch开发的核心思维在深度学习技术日新月异的今天,掌握PyTorch不仅仅意味着能够搭建和训练神经网络,更重要的是理解如何高效地利用现代硬件资源、优化模型性能并构建可扩展的AI系统。随着PyTorch2.x系列的成熟,特别是最新2.7版本的发布,框架为开发者提供了前所未有的优化工具和性能潜力。本文将深入探讨现代PyTorch开发中的核心优化技能,从编译器优化到注意力机制革新,从内
- 大语言模型的具身化——LLM-based Agents实战
apollowin123
人工智能语言模型深度学习
1.概述1.1Agent是什么长期以来,研究者们一直在追求与人类相当、乃至超越人类水平的通用人工智能(ArtificialGeneralIntelligence,AGI)。早在1950年代,AlanTuring就将「智能」的概念扩展到了人工实体,并提出了著名的图灵测试。这些人工智能实体通常被称为——代理(Agent)。「代理」这一概念起源于哲学,描述了一种拥有欲望、信念、意图以及采取行动能力的实体
- Node.js 后台系统 - 基本增删改查实现
个人简介个人主页:魔术师学习方向:主攻前端方向,正逐渐往全栈发展个人状态:研发工程师,现效力于政务服务网事业人生格言:“心有多大,舞台就有多大。”推荐学习:Vue2Vue3Vue2/3项目实战Node.js实战Three.js鸿蒙开发小程序使用备注:仅供学习交流严禁用于商业用途,若发现侵权内容请及时联系作者更新进度:持续更新内容个人名片:每篇文章最下方都有加入方式,旨在交流学习&资源分享,快加
- ace.js在线代码编辑器实战
明月566
js在线代码编辑器js代码编辑器语法检测ace.jsace.js在线代码编辑器
背景ACE简介:功能实现1、引入js2、添加控件3、初始化组件4、保存时代码语法检测5、效果图及完整示例代码:6、官网在线测试:7、遇到的一些问题:背景项目需要,在一些场景,用户需要手动编写一些js脚本来实现自己的功能;前期一直用文本框显示,不便于编辑和查看。因此需要引入一个在线代码编辑器。效果如下:ACE简介:ACE是一个开源的、独立的、基于浏览器的代码编辑器,可以嵌入到任何web页面或Java
- 10.5 实战ChatGLM3私有数据微调之提示工程:批量生成数据稳定性秘籍
少林码僧
掌握先机!从0起步实战AI大模型微调打造核心竞争力机器学习深度学习人工智能语言模型
实战ChatGLM3私有数据微调之提示工程:批量生成数据稳定性秘籍在当今人工智能蓬勃发展的时代,大语言模型(LLMs)如ChatGLM3的出现,为自然语言处理领域带来了革命性的变化。企业和开发者们纷纷寻求利用这些强大的模型来构建定制化的应用,以满足特定业务需求。其中,使用私有数据对ChatGLM3进行微调,成为了实现差异化竞争和提供个性化服务的关键途径。然而,在微调过程中,确保批量生成数据的稳定性
- 10.6 ChatGLM3私有数据微调实战:24小时打造高精度模型,显存直降60%
少林码僧
掌握先机!从0起步实战AI大模型微调打造核心竞争力chatgpt机器学习深度学习人工智能语言模型
ChatGLM3私有数据微调实战:24小时打造高精度模型,显存直降60%1.实战构造私有的微调数据集在微调大模型时,数据质量直接决定模型效果。本节将手把手教你如何构建高质量的私有微调数据集。1.1使用ChatGPT自动设计生成训练数据的Prompt核心思路:通过ChatGPT生成符合任务需求的样本数据,降低人工标注成本。步骤示例(以生成客服对话数据为例):fromlangchain.prompts
- Mermaid 绘图指南(一)- Mermaid图表绘制语法详解与实战示例
全能骑士涛锅锅
通用技术/研究方法MarkdownMermaid图表绘制流程图
Mermaid绘图指南(一)-Mermaid图表绘制语法详解与实战示例AuthorDateVersionNoteTaoWang2025-04-24V1.0Releasethedocument.文章目录Mermaid绘图指南(一)-Mermaid图表绘制语法详解与实战示例一、工具概述1.1工具简介1.2快速入门1.3图表类型支持1.4实战案例:明代皇族谱系二、饼状图开发规范2.1语法结构参数说明表2
- 领域驱动设计精要
MoneyHacksPro
Java场景面试宝典DDDDomain-DrivenDesignSoftwareArchitecture
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- 领域驱动设计实践精要
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- Spring框架核心技术解析
MoneyHacksPro
Java场景面试宝典SpringFrameworkDependencyInjectionAOP
我是廖志伟,一名Java开发工程师、《Java项目实战——深入理解大型互联网企业通用技术》(基础篇)、(进阶篇)、(架构篇)清华大学出版社签约作家、Java领域优质创作者、CSDN博客专家、阿里云专家博主、51CTO专家博主、产品软文专业写手、技术文章评审老师、技术类问卷调查设计师、幕后大佬社区创始人、开源项目贡献者。拥有多年一线研发和团队管理经验,研究过主流框架的底层源码(Spring、Spri
- Dom
周华华
JavaScripthtml
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/1999/xhtml&q
- 【Spark九十六】RDD API之combineByKey
bit1129
spark
1. combineByKey函数的运行机制
RDD提供了很多针对元素类型为(K,V)的API,这些API封装在PairRDDFunctions类中,通过Scala隐式转换使用。这些API实现上是借助于combineByKey实现的。combineByKey函数本身也是RDD开放给Spark开发人员使用的API之一
首先看一下combineByKey的方法说明:
- msyql设置密码报错:ERROR 1372 (HY000): 解决方法详解
daizj
mysql设置密码
MySql给用户设置权限同时指定访问密码时,会提示如下错误:
ERROR 1372 (HY000): Password hash should be a 41-digit hexadecimal number;
问题原因:你输入的密码是明文。不允许这么输入。
解决办法:用select password('你想输入的密码');查询出你的密码对应的字符串,
然后
- 路漫漫其修远兮 吾将上下而求索
周凡杨
学习 思索
王国维在他的《人间词话》中曾经概括了为学的三种境界古今之成大事业、大学问者,罔不经过三种之境界。“昨夜西风凋碧树。独上高楼,望尽天涯路。”此第一境界也。“衣带渐宽终不悔,为伊消得人憔悴。”此第二境界也。“众里寻他千百度,蓦然回首,那人却在灯火阑珊处。”此第三境界也。学习技术,这也是你必须经历的三种境界。第一层境界是说,学习的路是漫漫的,你必须做好充分的思想准备,如果半途而废还不如不要开始。这里,注
- Hadoop(二)对话单的操作
朱辉辉33
hadoop
Debug:
1、
A = LOAD '/user/hue/task.txt' USING PigStorage(' ')
AS (col1,col2,col3);
DUMP A;
//输出结果前几行示例:
(>ggsnPDPRecord(21),,)
(-->recordType(0),,)
(-->networkInitiation(1),,)
- web报表工具FineReport常用函数的用法总结(日期和时间函数)
老A不折腾
finereport报表工具web开发
web报表工具FineReport常用函数的用法总结(日期和时间函数)
说明:凡函数中以日期作为参数因子的,其中日期的形式都必须是yy/mm/dd。而且必须用英文环境下双引号(" ")引用。
DATE
DATE(year,month,day):返回一个表示某一特定日期的系列数。
Year:代表年,可为一到四位数。
Month:代表月份。
- c++ 宏定义中的##操作符
墙头上一根草
C++
#与##在宏定义中的--宏展开 #include <stdio.h> #define f(a,b) a##b #define g(a) #a #define h(a) g(a) int main() { &nbs
- 分析Spring源代码之,DI的实现
aijuans
springDI现源代码
(转)
分析Spring源代码之,DI的实现
2012/1/3 by tony
接着上次的讲,以下这个sample
[java]
view plain
copy
print
- for循环的进化
alxw4616
JavaScript
// for循环的进化
// 菜鸟
for (var i = 0; i < Things.length ; i++) {
// Things[i]
}
// 老鸟
for (var i = 0, len = Things.length; i < len; i++) {
// Things[i]
}
// 大师
for (var i = Things.le
- 网络编程Socket和ServerSocket简单的使用
百合不是茶
网络编程基础IP地址端口
网络编程;TCP/IP协议
网络:实现计算机之间的信息共享,数据资源的交换
协议:数据交换需要遵守的一种协议,按照约定的数据格式等写出去
端口:用于计算机之间的通信
每运行一个程序,系统会分配一个编号给该程序,作为和外界交换数据的唯一标识
0~65535
查看被使用的
- JDK1.5 生产消费者
bijian1013
javathread生产消费者java多线程
ArrayBlockingQueue:
一个由数组支持的有界阻塞队列。此队列按 FIFO(先进先出)原则对元素进行排序。队列的头部 是在队列中存在时间最长的元素。队列的尾部 是在队列中存在时间最短的元素。新元素插入到队列的尾部,队列检索操作则是从队列头部开始获得元素。
ArrayBlockingQueue的常用方法:
- JAVA版身份证获取性别、出生日期及年龄
bijian1013
java性别出生日期年龄
工作中需要根据身份证获取性别、出生日期及年龄,且要还要支持15位长度的身份证号码,网上搜索了一下,经过测试好像多少存在点问题,干脆自已写一个。
CertificateNo.java
package com.bijian.study;
import java.util.Calendar;
import
- 【Java范型六】范型与枚举
bit1129
java
首先,枚举类型的定义不能带有类型参数,所以,不能把枚举类型定义为范型枚举类,例如下面的枚举类定义是有编译错的
public enum EnumGenerics<T> { //编译错,提示枚举不能带有范型参数
OK, ERROR;
public <T> T get(T type) {
return null;
- 【Nginx五】Nginx常用日志格式含义
bit1129
nginx
1. log_format
1.1 log_format指令用于指定日志的格式,格式:
log_format name(格式名称) type(格式样式)
1.2 如下是一个常用的Nginx日志格式:
log_format main '[$time_local]|$request_time|$status|$body_bytes
- Lua 语言 15 分钟快速入门
ronin47
lua 基础
-
-
单行注释
-
-
[[
[多行注释]
-
-
]]
-
-
-
-
-
-
-
-
-
-
-
1.
变量 & 控制流
-
-
-
-
-
-
-
-
-
-
num
=
23
-
-
数字都是双精度
str
=
'aspythonstring'
- java-35.求一个矩阵中最大的二维矩阵 ( 元素和最大 )
bylijinnan
java
the idea is from:
http://blog.csdn.net/zhanxinhang/article/details/6731134
public class MaxSubMatrix {
/**see http://blog.csdn.net/zhanxinhang/article/details/6731134
* Q35
求一个矩阵中最大的二维
- mongoDB文档型数据库特点
开窍的石头
mongoDB文档型数据库特点
MongoDD: 文档型数据库存储的是Bson文档-->json的二进制
特点:内部是执行引擎是js解释器,把文档转成Bson结构,在查询时转换成js对象。
mongoDB传统型数据库对比
传统类型数据库:结构化数据,定好了表结构后每一个内容符合表结构的。也就是说每一行每一列的数据都是一样的
文档型数据库:不用定好数据结构,
- [毕业季节]欢迎广大毕业生加入JAVA程序员的行列
comsci
java
一年一度的毕业季来临了。。。。。。。。
正在投简历的学弟学妹们。。。如果觉得学校推荐的单位和公司不适合自己的兴趣和专业,可以考虑来我们软件行业,做一名职业程序员。。。
软件行业的开发工具中,对初学者最友好的就是JAVA语言了,网络上不仅仅有大量的
- PHP操作Excel – PHPExcel 基本用法详解
cuiyadll
PHPExcel
导出excel属性设置//Include classrequire_once('Classes/PHPExcel.php');require_once('Classes/PHPExcel/Writer/Excel2007.php');$objPHPExcel = new PHPExcel();//Set properties 设置文件属性$objPHPExcel->getProperties
- IBM Webshpere MQ Client User Issue (MCAUSER)
darrenzhu
IBMjmsuserMQMCAUSER
IBM MQ JMS Client去连接远端MQ Server的时候,需要提供User和Password吗?
答案是根据情况而定,取决于所定义的Channel里面的属性Message channel agent user identifier (MCAUSER)的设置。
http://stackoverflow.com/questions/20209429/how-mca-user-i
- 网线的接法
dcj3sjt126com
一、PC连HUB (直连线)A端:(标准568B):白橙,橙,白绿,蓝,白蓝,绿,白棕,棕。 B端:(标准568B):白橙,橙,白绿,蓝,白蓝,绿,白棕,棕。 二、PC连PC (交叉线)A端:(568A): 白绿,绿,白橙,蓝,白蓝,橙,白棕,棕; B端:(标准568B):白橙,橙,白绿,蓝,白蓝,绿,白棕,棕。 三、HUB连HUB&nb
- Vimium插件让键盘党像操作Vim一样操作Chrome
dcj3sjt126com
chromevim
什么是键盘党?
键盘党是指尽可能将所有电脑操作用键盘来完成,而不去动鼠标的人。鼠标应该说是新手们的最爱,很直观,指哪点哪,很听话!不过常常使用电脑的人,如果一直使用鼠标的话,手会发酸,因为操作鼠标的时候,手臂不是在一个自然的状态,臂肌会处于绷紧状态。而使用键盘则双手是放松状态,只有手指在动。而且尽量少的从鼠标移动到键盘来回操作,也省不少事。
在chrome里安装 vimium 插件
- MongoDB查询(2)——数组查询[六]
eksliang
mongodbMongoDB查询数组
MongoDB查询数组
转载请出自出处:http://eksliang.iteye.com/blog/2177292 一、概述
MongoDB查询数组与查询标量值是一样的,例如,有一个水果列表,如下所示:
> db.food.find()
{ "_id" : "001", "fruits" : [ "苹
- cordova读写文件(1)
gundumw100
JavaScriptCordova
使用cordova可以很方便的在手机sdcard中读写文件。
首先需要安装cordova插件:file
命令为:
cordova plugin add org.apache.cordova.file
然后就可以读写文件了,这里我先是写入一个文件,具体的JS代码为:
var datas=null;//datas need write
var directory=&
- HTML5 FormData 进行文件jquery ajax 上传 到又拍云
ileson
jqueryAjaxhtml5FormData
html5 新东西:FormData 可以提交二进制数据。
页面test.html
<!DOCTYPE>
<html>
<head>
<title> formdata file jquery ajax upload</title>
</head>
<body>
<
- swift appearanceWhenContainedIn:(version1.2 xcode6.4)
啸笑天
version
swift1.2中没有oc中对应的方法:
+ (instancetype)appearanceWhenContainedIn:(Class <UIAppearanceContainer>)ContainerClass, ... NS_REQUIRES_NIL_TERMINATION;
解决方法:
在swift项目中新建oc类如下:
#import &
- java实现SMTP邮件服务器
macroli
java编程
电子邮件传递可以由多种协议来实现。目前,在Internet 网上最流行的三种电子邮件协议是SMTP、POP3 和 IMAP,下面分别简单介绍。
◆ SMTP 协议
简单邮件传输协议(Simple Mail Transfer Protocol,SMTP)是一个运行在TCP/IP之上的协议,用它发送和接收电子邮件。SMTP 服务器在默认端口25上监听。SMTP客户使用一组简单的、基于文本的
- mongodb group by having where 查询sql
qiaolevip
每天进步一点点学习永无止境mongo纵观千象
SELECT cust_id,
SUM(price) as total
FROM orders
WHERE status = 'A'
GROUP BY cust_id
HAVING total > 250
db.orders.aggregate( [
{ $match: { status: 'A' } },
{
$group: {
- Struts2 Pojo(六)
Luob.
POJOstrust2
注意:附件中有完整案例
1.采用POJO对象的方法进行赋值和传值
2.web配置
<?xml version="1.0" encoding="UTF-8"?>
<web-app version="2.5"
xmlns="http://java.sun.com/xml/ns/javaee&q
- struts2步骤
wuai
struts
1、添加jar包
2、在web.xml中配置过滤器
<filter>
<filter-name>struts2</filter-name>
<filter-class>org.apache.st