这篇文章可以说是怎么分析和展示RNAseq基因表达数据中基因的相关性的延续。上次绘制了下图:
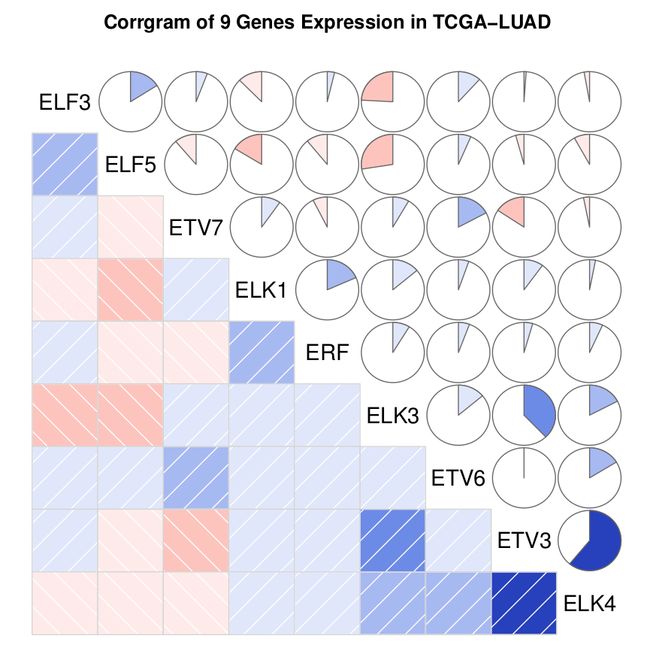
可以发现只有两个基因的表达表现出了较强的相关(ETV3-ELK4)。
一般教材描述相关性大小为:
相关系数r 是否是:·|r|>0.95 存在显著性相关;
·|r|≥0.8 高度相关;
·0.5≤|r|<0.8 中度相关;
·0.3≤|r|<0.5 低度相关;
·|r|<0.3 关系极弱,认为不相关
计算公式为:
 公式
公式
可见这两个基因属于中度相关。
但是我们知道皮尔逊相关系数表示的是两组数据线性相关的程度,但是如果两者在统计学上不存在相关性呢?那这个指标还有什么意义?因此,我们在评判相关的时候需要同时考量p值和r相关系数大小。
一个博主是这样认为的:
看两者是否算相关要看两方面:显著水平以及相关系数
(1)显著水平,就是P值,这是首要的,因为如果不显著,相关系数再高也没用,可能只是因为偶然因素引起的,那么多少才算显著,一般p值小于0.05就是显著了;如果小于0.01就更显著;例如p值=0.001,就是很高的显著水平了,只要显著,就可以下结论说:拒绝原假设无关,两组数据显著相关也说两者间确实有明显关系.通常需要p值小于0.1,最好小于0.05设甚至0.01,才可得出结论:两组数据有明显关系,如果p=0.5,远大于0.1,只能说明相关程度不明显甚至不相关.起码不是线性相关.
(2)相关系数,也就是Pearson Correlation(皮尔逊相关系数),通常也称为R值,在确认上面指标显著情况下,再来看这个指标,一般相关系数越高表明两者间关系越密切.
在搜索相关概念时,发现百度文库一篇文档相关系数与P值的一些基本概念提供了详细的描述和实例。有兴趣可以看下。
从上面看来,在进行相关分析考量相关系数r(或者R2)前,先考量显著性是有必要的。不过,如果你已经能看到两变量有很明显的线性关系了,你不看p值也无伤大雅,因为那个时候p值肯定少于0.05。
参考R包corrplot文档对上次的函数进行优化,代码如下:
gene_exp.corr <- function(gene.list, project_code, project.clinical, project.exp, outdir, ID_transform=TRUE, conf.level=0.95){
# Arguments:
# gene.list: a list of gene you want to analyze their expression correlation
# project_code: data project name or name you want to specify this analysis
# project.clinical: clinical information about samples, data.frame format
# project.exp: normalized gene expression (RNA seq) about samples, data.frame format
# ID_transform: sometimes clinical information use "-" as separate symbol for sample ID,
# we need it to be the same as it in project.exp data
# one sample ID example: in clinical information, one sample may be marked by "TCGA-3N-A9WB-06",
# in RNA seq data.set, this sample is "TCGA.3N.A9WB.06". If it is not, set ID_transform=FALSE.
# note: you need to install "corrgram" package before use this function
gene_exp.list <- subset(project.exp, sample%in%gene.list)
rownames(gene_exp.list) <- gene_exp.list[,1]
gene_exp.list <- gene_exp.list[,-1]
gene_exp.list <- t(gene_exp.list)
# gene_exp.list <- gene_exp.list[,c(5,1,2,3,4,6,7,8,9,10)]
library(corrplot)
# combine with significance test
cor.mtest <- function(mat, conf.level = 0.95){
mat <- as.matrix(mat)
n <- ncol(mat)
p.mat <- lowCI.mat <- uppCI.mat <- matrix(NA, n, n)
diag(p.mat) <- 0
diag(lowCI.mat) <- diag(uppCI.mat) <- 1
for(i in 1:(n-1)){
for(j in (i+1):n){
tmp <- cor.test(mat[,i], mat[,j], conf.level = conf.level)
p.mat[i,j] <- p.mat[j,i] <- tmp$p.value
lowCI.mat[i,j] <- lowCI.mat[j,i] <- tmp$conf.int[1]
uppCI.mat[i,j] <- uppCI.mat[j,i] <- tmp$conf.int[2]
}
}
return(list(p.mat, lowCI.mat, uppCI.mat))
}
if(ID_transform){
project.clinical$sampleID = gsub("-",".",project.clinical$sampleID, fixed = TRUE)
}
n.gene <- ncol(gene_exp.list)
# all samples
M1 <- cor(gene_exp.list)
res1 <- cor.mtest(gene_exp.list, conf.level)
pdf(paste(outdir,project_code,"_all_sample_genelist_expression_corrgram.pdf", sep=""))
corrplot(M1, order = "AOE", tl.pos = "d", p.mat = res1[[1]], insig = "p-value")
title(paste("Corrgram of ", n.gene," Genes Expression in ", project_code, sep = ""))
dev.off()
# choose tumor sample
# table(project.clinical$sample_type)
primary_tumor <- "Primary Tumor"
Metast_tumor <- "Metastatic"
primary_tumor.id <- project.clinical[project.clinical$sample_type==primary_tumor,]$sampleID
Metast_tumor.id <- project.clinical[project.clinical$sample_type==Metast_tumor,]$sampleID
if(length(primary_tumor.id)<2 & length(Metast_tumor.id<2)){
stop("Maybe your data have something wrong. Please check it!")
}else{
if(length(primary_tumor.id)<2){
stop("I don't think it's reasonable that there are less than 2 primary tumor samples.")}
gene_exp.list.primary <- subset(gene_exp.list, rownames(gene_exp.list)%in%primary_tumor.id)
M2 <- cor(gene_exp.list.primary)
res2 <- cor.mtest(gene_exp.list.primary, conf.level)
pdf(paste(outdir,project_code,"_primary_tumor_sample_genelist_expression_corrgram.pdf", sep=""))
corrplot(M2, order = "AOE", tl.pos = "d", p.mat = res2[[1]], insig = "p-value")
title(paste("Corrgram of ", n.gene," Genes Expression in ", project_code, sep = ""))
dev.off()
if(length(Metast_tumor.id)<2){
cat("It seems has no Metastatic sample in this analysis. \n")
return(0)
}
gene_exp.list.Metast <- subset(gene_exp.list, rownames(gene_exp.list)%in%Metast_tumor.id)
M3 <- cor(gene_exp.list.Metast)
res3 <- cor.mtest(gene_exp.list.Metast, conf.level)
pdf(paste(outdir,project_code,"_Metastatic_sample_genelist_expression_corrgram.pdf", sep=""))
corrplot(M3, order = "AOE", tl.pos = "d", p.mat = res3[[1]], insig = "p-value")
title(paste("Corrgram of ", n.gene," Genes Expression in ", project_code, sep = ""))
dev.off()}
}
一方面增加了检验部分,另一方面修改了画图函数。如果你想用这个函数绘制更多自定义的图,可以参考R包文档进行修改。
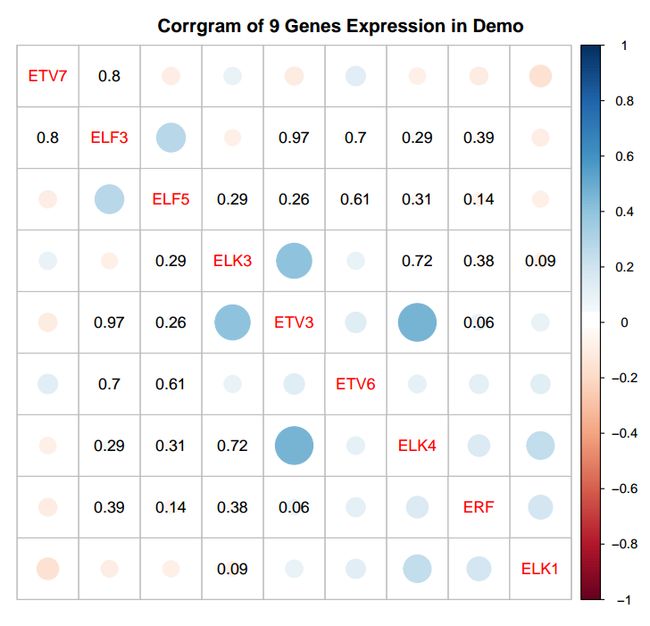
这跟上次的图类似。偏蓝色代表正相关,偏红色代表负相关。用圆圈大小和颜色鲜艳程度辅助color legend可以很好的区分和找出相关性明显的。corrgram的左下角和右上角是对称的,标有数字的显示的是p值,我这里默认设定0.05为阈值,大于0.05都会显示出来,这些值说明对应的两个基因在统计学上是没有相关性的。
博文链接