seqtk安装和使用
------------------------------
Nickier
2018-12-13
------------------------------
1.下载
conda search seqtk
conda install -y seqtk
2.查看seqtk的子命令
$seqtk
Usage: seqtk
Version: 1.3-r106
Command: seq common transformation of FASTA/Q # FASTA/Q 的转换
comp get the nucleotide composition of FASTA/Q # 获取FASTA/Q的核苷酸组成
sample subsample sequences # 获取样本序列
subseq extract subsequences from FASTA/Q # 提取子序列
fqchk fastq QC (base/quality summary) # fastq的质控
mergepe interleave two PE FASTA/Q files #交叉合并双端测序的两个FASTA/Q files,合并后的file第一条序列是第一个fq的第一条,合并后的file第二条是序列是第二个fq的第一条
trimfq trim FASTQ using the Phred algorithm # 用Phred算法对fq修剪
hety regional heterozygosity # 区域性杂合
gc identify high- or low-GC regions# 识别高低GC含量的区域
mutfa point mutate FASTA at specified positions#在特定位置指出FASTA的突变
mergefa merge two FASTA/Q files # 合并两个的FASTA/Q files
famask apply a X-coded FASTA to a source FASTA#将X编码的fa应用到原fa
dropse drop unpaired from interleaved PE FASTA/Q# 从交错合并的fa/fq中丢弃不成对的序列
rename rename sequence names#序列重命名
randbase choose a random base from hets#从hets中随机选一个碱基
cutN cut sequence at long N # 在N长度处切掉序列
listhet extract the position of each het #提取每一个het位置
3.命令
3.1 seqtk seq
$seqtk seq
Usage: seqtk seq [options] |
Options: -q INT mask bases with quality lower than INT [0] #将质量小于INT的碱基,转成小写的字母[atgc],INT默认是0。
-X INT mask bases with quality higher than INT [255] #标记质量大于INT的碱基。INT默认是255
-n CHAR masked bases converted to CHAR; 0 for lowercase [0]#用给出的CHAR字符代替标记的碱基,0表示用原碱基的小写字母表示。默认为0。这个参数应该和-q 或-X 搭配使用
-l INT number of residues per line; 0 for 2^32-1 [0] #每条序列取前INT个碱基。默认为0,表示保留整条序列
-Q INT quality shift: ASCII-INT gives base quality [33]#质量变换,如ASCII-33
-s INT random seed (effective with -f) [11]#随机种子,配合-f参数,默认11
-f FLOAT sample FLOAT fraction of sequences [1]#随机取整个文件的FLOAT(例如:0.5)行,随机数种子由-s决定。默认为1,表示保留所有序列
-M FILE mask regions in BED or name list FILE [null]#FILE可以是BED文件。若是BED文件,就将BED文件中给定区间的碱基转换成小写[atgc]序列;若是列表文件,则屏蔽掉给定的ID对应的整条序列。默认为空
-L INT drop sequences with length shorter than INT [0]#丢弃掉长度小于INT的序列,默认是0
-F CHAR fake FASTQ quality []# 用CHAR字符替换fq的质量值
-c mask complement region (effective with -M)# 标记互补区域,和-M参数配合使用
-r reverse complement#生成反向互补序列
-A force FASTA output (discard quality)#强制转换成fa格式,也可以用-a
-C drop comments at the header lines#在标题行删除注释
-N drop sequences containing ambiguous bases#丢弃掉含有不确定碱基N的序列
-1 output the 2n-1 reads only#仅输入2n-1(奇数)的reads
-2 output the 2n reads only#仅输入2n-1(偶数)的reads
-V shift quality by '(-Q) - 33' #通过-Q-33转换质量值
-U convert all bases to uppercases#所有碱基换成大写字母
-S strip of white spaces in sequences#删除序列中的空白行
-q将质量小于INT的碱基,转成小写的字母[atgc],INT默认是0。
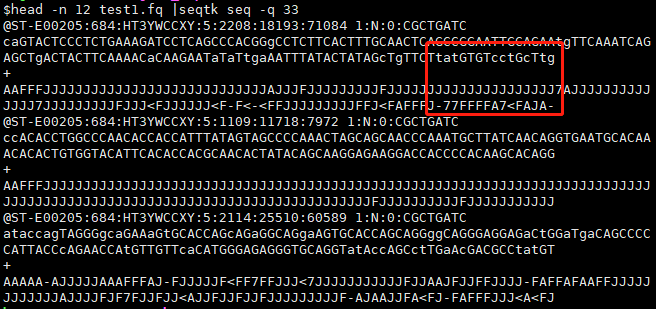
-q.png
-q 和-n
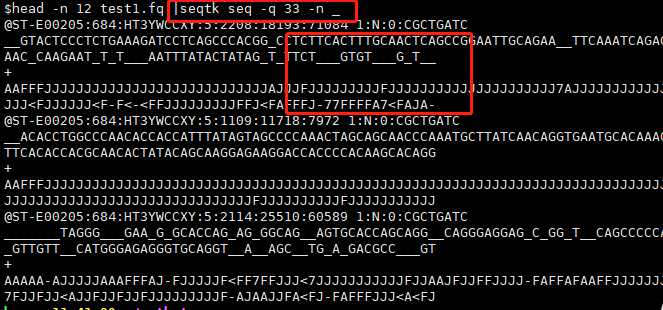
-n.png
-r生成反向互补序列

-r.png
3.2 seqtk comp
$seqtk comp
Usage: seqtk comp [-u] [-r in.bed] # 获取fa的碱基的组成信息,用-r参数可以输出bed中的给定区间的序列
Output format: chr, length, #A, #C, #G, #T, #2, #3, #4, #CpG, #tv, #ts, #CpG-ts
# 输出格式:序列号 序列长度 A C G T

comp.png
3.3 seqtk sample
$seqtk sample
Usage: seqtk sample [-2] [-s seed=11] |
#随机抽取序列,用法是seqtk sample fq/fa num
Options: -s INT RNG seed [11]#设置随机种子,默认11
-2 2-pass mode: twice as slow but with much reduced memory#占用更大的内存
3.4 seqtk subseq
$seqtk subseq
Usage: seqtk subseq [options] |
#提取name.list中指定名称的fa序列,
Options: -t TAB delimited output# 输出以tab分割
-l INT sequence line length [0]# 输出序列以长度INT换行
Note: Use 'samtools faidx' if only a few regions are intended.#注意:如果只有少数几个区域,请使用'samtools faidx'

subseq.png
3.5 seqtk fqchk
$seqtk fqchk
Usage: seqtk fqchk [-q 20] #获取每个碱基点的分布和质量值,和fastqc质控类似,不过这里生成的是数据,而fastqc生成质控报告
Note: use -q0 to get the distribution of all quality values#用-q0来获取所有质量值的分布

fqchk.png
3.6 seqtk mergepe
$seqtk mergepe
Usage: seqtk mergepe
# 交叉合并双端测序的序列,pe就是pair end的意思
3.7 seqtk trimfq
$seqtk trimfq
Usage: seqtk trimfq [options]
Options: -q FLOAT error rate threshold (disabled by -b/-e) [0.05]#设置错误率的阈值为FLOAT,以此作为修剪标准。此参数不可与-b/-e参数同时使用。默认值为0.05
-l INT maximally trim down to INT bp (disabled by -b/-e) [30]#无论是否质量低,序列保留到至少INT长度。此参数不可与-b/-e参数同时使用。默认值为30。此参数可以看下图(R2.fastq有三条read,测序质量依次递增)
-b INT trim INT bp from left (non-zero to disable -q/-l) [0]# 从序列左边切除INT个碱基。此参数不可与-q/-l参数同时使用。默认值为0
-e INT trim INT bp from right (non-zero to disable -q/-l) [0]#从序列右边切除INT个碱基。此参数不可与-q/-l参数同时使用。默认值为0
-L INT retain at most INT bp from the 5'-end (non-zero to disable -q/-l) [0]#保留从5'端起前INT个碱基
-Q force FASTQ output#强制输出fq格式

trimfq.png
3.8 seqtk hety
$seqtk hety
Usage: seqtk hety [options] #区域性杂合
Options: -w INT window size [50000]
-t INT # start positions in a window [5]
-m treat lowercases as masked
3.9 seqtk gc
$seqtk gc
Usage: seqtk gc [options] # 识别高低GC含量的区域
Options:
-w identify high-AT regions
-f FLOAT min GC fraction (or AT fraction for -w) [0.60]
-l INT min region length to output [20]
-x FLOAT X-dropoff [10.0]
3.10 seqtk mutfa
$seqtk mutfa
Usage: seqtk mutfa #给定in.snp特定位置指出fa的突变
Note: contains at least four columns per line which are:
'chr 1-based-pos any base-changed-to'.
3.11 seqtk mergefa
$seqtk mergefa
Usage: seqtk mergefa [options] # 合并两个的FASTA/Q files
Options: -q INT quality threshold [0]
-i take intersection#取交集
-m convert to lowercase when one of the input base is N
-r pick a random allele from het
-h suppress hets in the input
3.12 seqtk famask
$seqtk famask# 将X编码的fa应用到原fa
Usage: seqtk famask
3.13 seqtk dropse
$seqtk dropse# 从交叉合并的fa/fq中丢弃不成对的序列
Usage: seqtk dropse
3.14 seqtk rename
$seqtk rename #序列重命名
Usage: seqtk rename [prefix]
3.15 seqtk randbase
$seqtk randbase#从hets中随机选一个碱基
Usage: seqtk randbase
3.16 seqtk cutN
$seqtk cutN# 在N长度处切掉序列
Usage: seqtk cutN [options]
Options: -n INT min size of N tract [1000]
-p INT penalty for a non-N [10]
-g print gaps only, no sequence
3.17 seqtk listhet
$seqtk listhet #提取每一个het位置
Usage: seqtk listhet