刘小泽写于2020.1.23
遇到的问题
就是常规的差异分析结果进行GO富集分析,代码是:
# 先获得差异分析结果DEG_mtx,然后按照log2FC排序
geneList <- DEG_mtx$log2FoldChange
# 这一步是去掉Ensemble ID的版本号,因为后面进行ID转换时,Ensemble ID是不支持版本号的
names(geneList) <- str_split(rownames(DEG_mtx) ,
pattern = '\\.',simplify = T)[,1]
geneList <- sort(geneList,decreasing = T)
得到了这个背景基因集(universe gene list)之后,就可以进行富集分析
library(org.Hs.eg.db)
up_BP <- enrichGO(gene = up.gene,
universe = names(geneList),
keyType = 'ENSEMBL',
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 1,
qvalueCutoff = 1,
readable = TRUE)
# 如果不用参数readable= TRUE,也可以使用设置up_BP富集分析结果的基因显示为Symbol ID
up_BP <- setReadable(up_BP, OrgDb = org.Hs.eg.db)
tmp <- as.data.frame(up_BP)
比如我关注:GO:0030154 (cell differentiation)这个通路,放在数据框中查找,结果却是空的

思考
按说这个通路应该是很大的,不应该找不到这个通路的。
首先查询这个通路
注意这个链接的特点,以后可以快速查找: https://www.ebi.ac.uk/QuickGO/term/GO:0030154
看到这个图,它也的确是在子节点,属于BP通路:
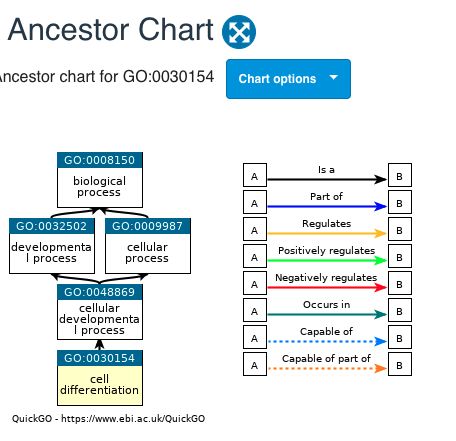
那么这个通路到底有多少基因呢?
刚开始想从网页直接查询,但几分钟都没有查到。然后突然想到,不是有org.db这个强大的R数据库吗,而且之前也写过如果去获取其中的信息:如何快速理解GO富集结果?
library(org.Hs.eg.db)
go2gene <- toTable(org.Hs.egGO2ALLEGS)
uni_gene <- unique(go2gene$gene_id[go2gene$go_id == 'GO:0030154'])
> length(uni_gene)
[1] 4239
这个通路竟然有4000多个基因
我们的差异基因有多少在这个通路呢?
# 我们总共有这些基因,但它们是Ensemble ID,而uni_gene是Entrez ID,需要转换一下
> length(up.gene)
[1] 1445
trans_id <- bitr(up.gene,fromType = "ENSEMBL",toType = "ENTREZID",
org.Hs.eg.db)
然后找二者交集
> length(intersect(trans_id$ENTREZID,uni_gene))
[1] 231
发现虽然我们这个通路有4000多个基因,但我们的1000多个差异基因中,也有200多个属于这个通路。因此不会不存在这个通路的
那么问题出在哪里?
我又看了一下enrichGO的参数,终于发现,原来它对GO term的基因数量做了限制:

它默认使用基因数量10-500的term进行富集分析,而我们这里的这个通路,由于基因数量4000多,所以不被纳入考量
因此,如果真的要看这个通路,那么只需要修改一下参数:
up_BP2 <- enrichGO(gene = up.gene,
universe = names(geneList),
keyType = 'ENSEMBL',
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 1,
qvalueCutoff = 1,
readable = TRUE,
minGSSize = 10, maxGSSize = 5000)
tmp2 <- as.data.frame(up_BP2)
也可以看到结果比之前多了几百个通路富集【当然这里没进行任何的过滤,因为可以事后再过滤】

然后就能看到,这个通路出现了,并且还很比较显著:

关于事后过滤
这个Y叔早已经多次介绍过了,并且很好用:https://wemp.app/posts/8ee2f29e-107d-4844-9c5c-7577ea3a1105
不过需要使用enrichplot的开发版
它的目的就是输出的富集分析结果作为数据框,然后自己过滤,最后把结果再转换为enrichResult对象,进行后续作图,例如:
x=up_BP
y = x[x$pvalue < 0.05, ]
> dim(x);dim(y)
[1] 4692 9
[1] 549 9
> class(y)
[1] "data.frame"
然后只需要加个参数:asis=T
y = x[x$pvalue < 0.05, asis=T]
> class(y)
[1] "enrichResult"
attr(,"package")
[1] "DOSE"
这样就完成了自行过滤,并且保持了原来的数据格式
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
