刘小泽写于2020.7.22
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合
单细胞交响乐1-常用的数据结构SingleCellExperiment
单细胞交响乐2-scRNAseq从实验到下游简介
单细胞交响乐3-细胞质控
单细胞交响乐4-归一化
单细胞交响乐5-挑选高变化基因
单细胞交响乐6-降维
单细胞交响乐7-聚类分群
单细胞交响乐8-marker基因检测
单细胞交响乐9-细胞类型注释
单细胞交响乐9-细胞类型注释
单细胞交响乐10-数据集整合后的批次矫正
单细胞交响乐11-多样本间差异分析
单细胞交响乐12-检测Doublet
单细胞交响乐13-细胞周期推断
单细胞交响乐14-细胞轨迹推断
单细胞交响乐15-scRNA与蛋白丰度信息结合
单细胞交响乐16-处理大型数据
单细胞交响乐17-不同单细胞R包的数据格式相互转换
单细胞交响乐18-实战一 Smart-seq2
单细胞交响乐19-实战二 STRT-Seq
单细胞交响乐20-实战三 10X 未过滤的PBMC数据
单细胞交响乐21-实战三 批量处理并整合多个10X PBMC数据
单细胞交响乐22-实战五 CEL-seq2
单细胞交响乐23-实战六 CEL-seq
单细胞交响乐24-实战七 SMARTer 胰腺细胞
单细胞交响乐25-实战八 Smart-seq2 胰腺细胞
单细胞交响乐26-实战九 胰腺细胞数据整合
单细胞交响乐27-实战十 CEL-seq-小鼠造血干细胞
单细胞交响乐28-实战十一 Smart-seq2-小鼠造血干细胞
单细胞交响乐29-实战十二 10X 小鼠嵌合体胚胎
单细胞交响乐30-实战十三 10X 小鼠乳腺上皮细胞
1 前言
前面的种种都是作为知识储备,但是不实战还是记不住前面的知识
这是第十四个实战练习,不过这次的练习对电脑要求比较高
单细胞领域不得不提的HCA计划
参考:
官网:https://data.humancellatlas.org/
http://www.casisd.cn/zkcg/ydkb/kjqykb/2016/201612/201707/t20170703_4821952.html
https://baike.baidu.com/tashuo/browse/content?id=6937242830be0d08a0384772
http://med.china.com.cn/content/pid/134504/tid/1015
细胞类型多种多样
细胞是生命最基本的单位,一般人至少有37兆2000亿个细胞,但现有的研究一般是组织层面,对细胞内部机制理解的还不是很深入。人体内的细胞有多少种类?这是一个非常基础的问题。美国国立卫生研究院给出了一个答案:200种,包括像神经元细胞、心脏细胞、肌肉细胞等几大类。然而,如果你去问一个免疫学家,他会告诉你光免疫细胞就至少有200种;如果你去问一个专门研究T细胞的免疫学家,他会告诉你光T细胞就至少有200种。Regev说她自己光讲人体细胞种类就要花15分钟,并且一个细胞的种类可以不断地往下划分,亚型下面还有亚型,仅视网膜组织就至少包括100种不同种类的神经元。更棘手的是,有些种类的细胞会在一定条件下转化为其他种类的细胞,并且每个亚型的细胞会根据不同环境呈现出不同的状态。
细胞图谱的重要性
一个完整的人体细胞图谱将赋予我们每种细胞类型唯一的“身份证”,可以帮助解释不同类型的细胞如何协作并形成组织的。另外与疾病相关的基因在哪些细胞更活跃,不同细胞类型产生的机理又是怎样的呢?
HCA计划的开启
2016年Chan Zuckerberg Initiative(CZI)基金会计划在未来十年内投入30亿美元支持科学研究,其中最瞩目的就是“人类细胞图谱计划”(the Human Cell Atlas)【可与“人类基因组计划”相媲美】。2017年10月,HCA正式公布了首批拟资助的38个项目,将为40万亿个细胞绘制图谱,其中清华大学教授张学工负责的项目作为其中唯一一个由中国科学家承担的项目,从全球范围内征集的近500个项目中脱颖而出。
2018年3月8日,Sanger研究所在其官网宣布了人类发育细胞图谱计划(HDCA)的最新成果研究团队从捐赠的人类发育组织(包括肝脏、皮肤、肾脏、胎盘)中分离出超25万个细胞,并利用强大的单细胞基因组测序工具获取了人类早期发育,以及影响健康或者导致疾病的信息。HDCA是HCA的重要一部分,旨在构建参与人类发育的所有重要细胞的基因组参考图谱。该项目聚焦的其他主要领域包括,改善对血细胞如何形成,以及免疫系统如何发挥功能的理解。
另外,2018年,美国费城儿童医院和辛辛那提儿童医院领导的国际研究团队建议在HCA联盟中建立一个纵向的儿童研究分支,并在HCA白皮书中概述了儿童细胞图谱计划(Pediatric Cell Atlas,PCA),以在儿童健康和人类发展背景下,展开针对儿童独特生物学特征的跨学科研究。
HCA的主要内容
- 编目所有人体细胞类型(如免疫细胞、脑细胞)及其子类型;
- 绘制不同细胞类型在组织和人体中的分布图;
- 区分细胞状态(如免疫细胞在被病原体激活前后的状态);
- 捕捉细胞转换过程的关键特征(如干细胞细胞激活和分化);
- 追踪细胞谱系历史(如骨髓前体干细胞到功能性的红细胞)
数据准备
我们这里使用的数据是:10X技术得到的人类约380,000个骨髓细胞
数据链接:https://share.weiyun.com/JLvQvpHS
# 如果要自己下载
# library(HCAData)
# sce.bone <- HCAData('ica_bone_marrow')
load('sce.bone.new.RData')
sce.bone
# class: SingleCellExperiment
# dim: 33694 378000
# metadata(0):
# assays(1): counts
# rownames(33694): ENSG00000243485
# ENSG00000237613 ... ENSG00000277475
# ENSG00000268674
# rowData names(2): ID Symbol
# colnames(378000):
# MantonBM1_HiSeq_1-AAACCTGAGCAGGTCA-1
# MantonBM1_HiSeq_1-AAACCTGCACACTGCG-1 ...
# MantonBM8_HiSeq_8-TTTGTCATCTGCCAGG-1
# MantonBM8_HiSeq_8-TTTGTCATCTTGAGAC-1
# colData names(1): Barcode
# reducedDimNames(0):
# altExpNames(0):
数据初探
# 这里的数据大小41M
object.size(counts(sce.bone))
# 41741200 bytes
# 真实文件大小700多M
file.info(path(counts(sce.bone)))$size
# [1] 769046295
# 其中包含供体信息
head(sce.bone$Barcode)
# [1] "MantonBM1_HiSeq_1-AAACCTGAGCAGGTCA-1"
# [2] "MantonBM1_HiSeq_1-AAACCTGCACACTGCG-1"
# [3] "MantonBM1_HiSeq_1-AAACCTGCACCGGAAA-1"
# [4] "MantonBM1_HiSeq_1-AAACCTGCATAGACTC-1"
# [5] "MantonBM1_HiSeq_1-AAACCTGCATCGATGT-1"
# [6] "MantonBM1_HiSeq_1-AAACCTGCATTCCTGC-1"
# 提取供体信息
sce.bone$Donor <- sub("_.*", "", sce.bone$Barcode)
table(sce.bone$Donor)
#
# MantonBM1 MantonBM2 MantonBM3 MantonBM4 MantonBM5
# 48000 48000 48000 48000 48000
# MantonBM6 MantonBM7 MantonBM8
# 42000 48000 48000
ID转换
还是要获得染色体信息
library(EnsDb.Hsapiens.v86)
rowData(sce.bone)$Chr <- mapIds(EnsDb.Hsapiens.v86, keys=rownames(sce.bone),
column="SEQNAME", keytype="GENEID")
# 其中包括13个线粒体信息
table(grepl('MT',rowData(sce.bone)$Chr))
#
# FALSE TRUE
# 33681 13
整合行名
library(scater)
rownames(sce.bone) <- uniquifyFeatureNames(rowData(sce.bone)$ID,
names = rowData(sce.bone)$Symbol)
2 质控
使用线粒体信息进行过滤,并且因为数据量很大,可以调用多线程
library(BiocParallel)
# 调用8线程
bpp <- MulticoreParam(8)
start=Sys.time()
sce.bone <- unfiltered <- addPerCellQC(sce.bone, BPPARAM=bpp,
subsets=list(Mito=which(rowData(sce.bone)$Chr=="MT")))
end=Sys.time()
(end-start)
# Time difference of 2.167978 mins
过滤掉了6万多个细胞
colSums(as.matrix(qc))
# low_lib_size low_n_features
# 33997 42756
# high_subsets_Mito_percent discard
# 44105 61275
作图
unfiltered$discard <- qc$discard
gridExtra::grid.arrange(
plotColData(unfiltered, x="Donor", y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, x="Donor", y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, x="Donor", y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)
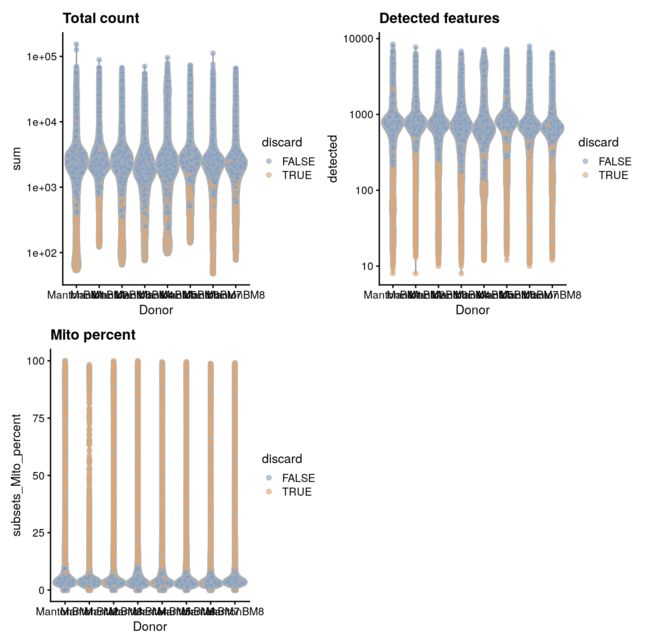
再看看线粒体含量与文库大小的关系
plotColData(unfiltered, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()
3 归一化
这里为了减少计算量,使用原来计算好的文库size factor,不需要重复计算
sce.bone <- logNormCounts(sce.bone, size_factors = sce.bone$sum)
summary(sizeFactors(sce.bone))
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.05 0.47 0.65 1.00 0.89 42.38
4 找高变异基因
将供体作为批次信息加入到构建模型中,这一步依然需要多线程处理
library(scran)
start=Sys.time()
dec.bone <- modelGeneVar(sce.bone, block=sce.bone$Donor, BPPARAM=bpp)
end=Sys.time()
(end-start) # Time difference of 10.58185 mins
top.bone <- getTopHVGs(dec.bone, n=5000)
5 矫正批次效应
library(batchelor)
library(BiocNeighbors)
set.seed(1010001)
merged.bone <- fastMNN(sce.bone, batch = sce.bone$Donor, subset.row = top.bone,
BSPARAM=BiocSingular::RandomParam(deferred = TRUE),
BNPARAM=AnnoyParam(),
BPPARAM=bpp)
reducedDim(sce.bone, 'MNN') <- reducedDim(merged.bone, 'corrected')
看下结果:lost.var 值越大表示丢失的真实生物异质性越多
## MantonBM1 MantonBM2 MantonBM3 MantonBM4 MantonBM5 MantonBM6 MantonBM7
## [1,] 0.010616 0.008241 0.000000 0.000000 0.000000 0.000000 0.000000
## [2,] 0.007894 0.007741 0.023662 0.000000 0.000000 0.000000 0.000000
## [3,] 0.005834 0.003823 0.005423 0.025272 0.000000 0.000000 0.000000
## [4,] 0.003482 0.002868 0.002581 0.003067 0.027117 0.000000 0.000000
## [5,] 0.005287 0.003544 0.003173 0.005702 0.006551 0.031890 0.000000
## [6,] 0.004601 0.004535 0.004533 0.004067 0.004934 0.005547 0.034452
## [7,] 0.002610 0.002238 0.003135 0.002799 0.001963 0.002697 0.002404
## MantonBM8
## [1,] 0.00000
## [2,] 0.00000
## [3,] 0.00000
## [4,] 0.00000
## [5,] 0.00000
## [6,] 0.00000
## [7,] 0.04033
6 降维聚类
降维
这么大的细胞数据,一般会使用UMAP
set.seed(01010100)
sce.bone <- runUMAP(sce.bone, dimred="MNN",
BNPARAM=AnnoyParam(),
BPPARAM=bpp,
n_threads=bpnworkers(bpp))
另外关于tSNE和UMAP的对比:可以看看https://towardsdatascience.com/how-exactly-umap-works-13e3040e1668
- SNE does not preserve global data structure, meaning that only within cluster distances are meaningful while between cluster similarities are not guaranteed. But UMAP Can Preserve Global Structure
- tSNE does not scale well for rapidly increasing sample sizes in scRNAseq.
- UMAP is faster than tSNE when it concerns a) large number of data points, b) number of embedding dimensions greater than 2 or 3, c) large number of ambient dimensions in the data set
- UMAP的改进:UMAP overall follows the philosophy of tSNE, but introduces a number of improvements such as another cost function and the absence of normalization of high- and low-dimensional probabilities.
- 总结:Despite tSNE served the Single Cell research area for years, it has too many disadvantages such as speed and the lack of global distance preservation
聚类
因为这里的细胞数量实在太大,因此可以先借用kmeans方法对它们聚类,比如先聚成1000个小类,然后再对每个小类进行graph-based聚类
set.seed(1000)
clust.bone <- clusterSNNGraph(sce.bone, use.dimred="MNN",
use.kmeans=TRUE, kmeans.centers=1000)
colLabels(sce.bone) <- factor(clust.bone)
table(colLabels(sce.bone))
##
## 1 2 3 4 5 6 7 8 9 10 11 12
## 21938 42850 19861 38064 38024 71978 19237 24627 7583 16361 3179 13023
做个热图
tab <- table(Cluster=colLabels(sce.bone), Donor=sce.bone$Donor)
library(pheatmap)
pheatmap(log10(tab+10), color=viridis::viridis(100))
看到每个cluster中都会有几个不同的样本,这也符合预期,因为所有的样本都是来自不同供体的重复,它们实际都是骨髓细胞

7 找marker基因
markers.bone <- findMarkers(sce.bone, block = sce.bone$Donor,
direction = 'up', lfc = 1, BPPARAM=bpp)
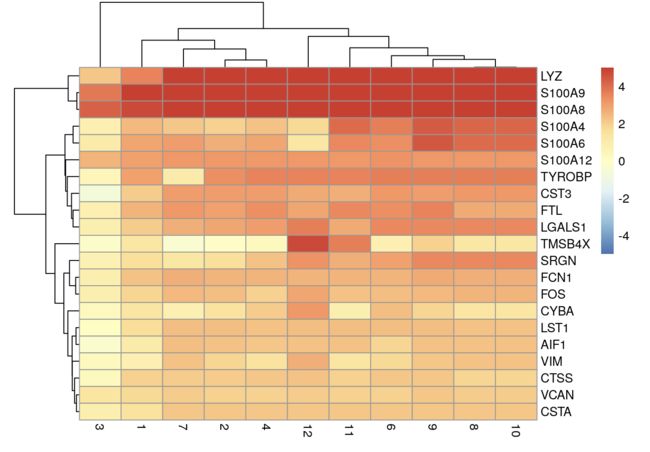
然后检查第5群细胞
top.markers <- markers.bone[["5"]]
best <- top.markers[top.markers$Top <= 10,]
lfcs <- getMarkerEffects(best)
library(pheatmap)
pheatmap(lfcs, breaks=seq(-5, 5, length.out=101))
看到LYZ、S100A8、VCAN基因高表达,说明cluster5可能是单核细胞(Monocyte)
8 细胞类型注释
使用参考数据集帮助注释,只不过这里并不是对每个细胞单独操作,因为数量太太。而是基于12个已经分好的cluster的总体表达量进行注释,提高了注释速度,但同样牺牲了研究cluster内部细胞异质性的机会。适用于快速注释细胞类群
se.aggregated <- sumCountsAcrossCells(sce.bone, id=colLabels(sce.bone))
library(SingleR)
hpc <- HumanPrimaryCellAtlasData()
anno.single <- SingleR(se.aggregated, ref = hpc, labels = hpc$label.main,
assay.type.test="sum")
看一下结果,包含12行也就是12个分群结果,看到利用参考注释的方法,cluster5也是被注释到了Monocyte这个细胞类型
anno.single
## DataFrame with 12 rows and 5 columns
## scores first.labels tuning.scores
##
## 1 0.388013:0.636789:0.754052:... CMP 0.554195:0.4238259
## 2 0.326563:0.692156:0.661226:... NK_cell 0.579208:0.4459558
## 3 0.323785:0.668599:0.743138:... Pre-B_cell_CD34- 0.488346:0.0719803
## 4 0.346204:0.637353:0.615021:... T_cells 0.628161:0.3531725
## 5 0.297369:0.638962:0.745831:... Pre-B_cell_CD34- 0.548995:0.2633979
## ... ... ... ...
## 8 0.313537:0.773897:0.672663:... B_cell 0.722126:-0.288616
## 9 0.346503:0.703506:0.670777:... Pro-B_cell_CD34+ 0.774848: 0.710191
## 10 0.313591:0.774162:0.668809:... B_cell 0.719013:-0.281128
## 11 0.369873:0.720021:0.654892:... B_cell 0.523979: 0.326187
## 12 0.403994:0.603811:0.697425:... MEP 0.430493: 0.286268
## labels pruned.labels
##
## 1 CMP CMP
## 2 T_cells T_cells
## 3 Monocyte Monocyte
## 4 T_cells T_cells
## 5 Monocyte Monocyte
## ... ... ...
## 8 B_cell B_cell
## 9 Pro-B_cell_CD34+ Pro-B_cell_CD34+
## 10 B_cell B_cell
## 11 B_cell NA
## 12 BM & Prog. BM & Prog.
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
