转发自http://crickcollege.com/news/97.html
上一篇关于质谱搜库的推文发出来后,很多小伙伴都发消息给我们说,常常被质谱鉴定中的各种统计学指标绕晕,希望能从我们这里重新找回方向…小编顿时有种莫名的责任感,于是特邀中科院计算所的高手,打算开个专题,详细跟大家聊一聊p值、E值、FDR、q值这些剪不断理还乱的玩意儿~ 要聊清楚不容易,篇幅所限,今天先开扒p值和E值,下一篇再扒FDR和q值!

背景铺垫
先介绍一下鸟枪法(Shotgun method),这真是一个很形象的名称,就像用霰弹枪打鸟,无论是针对基因测序技术,还是蛋白质谱技术,基本原理都是把长长的DNA/蛋白序列打散(酶切)成若干小片段,再通过检测技术弄清楚这些小片段的残基组成,最后再将这些片段的序列连接起来。
鸟枪法蛋白质组学,说得详细一些,就是基于串联质谱技术,先鉴定肽段,进而推断蛋白质序列。流程可以这样描述:
打散:通过酶切,把蛋白质切碎得到肽段混合物;
检测:肽段混合物经过色谱分离和离子化后,经串联质谱碎裂产生实验谱图;
鉴定:谱库搜索是目前最常用的鉴定方法[1, 2],就是拿实验谱图与数据库中的理论谱图进行匹配,得到可能的肽段序列。
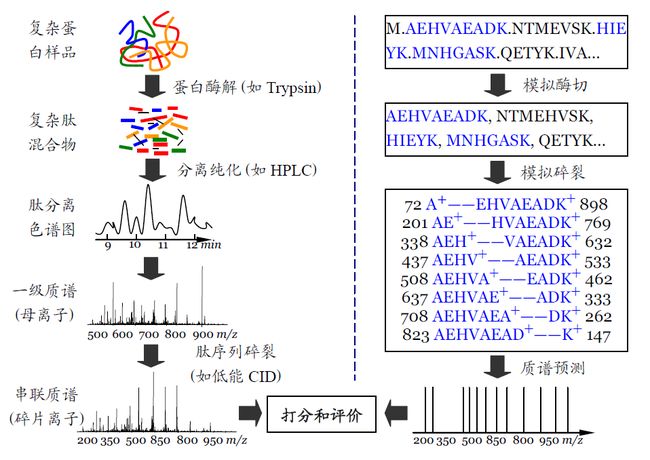
鸟枪法蛋白质组学流程
说到鉴定,就像足球的射门、篮球的投篮、排球的扣杀一样,是最后见分晓的一步!问题是,我们能保证鉴定的结果都是对的吗?
要讨论这个问题,我们首先来了解一下谱图匹配这种事儿具体是怎么玩的。
首先,我们得选一个蛋白序列数据库,可以是公共数据库,也可以是你自建的,要与你所研究的物种对应,通常还要加入实验室常见污染物数据库。如果你研究的物种还没有比较完整的蛋白序列数据库,那就选一类进化关系最近的物种的序列库。
例如目前最大最完整的蛋白数据库之一UniProt
图片来源:http://www.uniprot.org/
选好数据库以后,搜索引擎会对库里所有的蛋白序列进行理论酶切,得到肽段序列,再对肽段序列进行理论碎裂,形成理论谱图。
然后,用每一张导入搜索引擎的实验谱图与落入母离子质量误差窗口内的理论谱图进行匹配打分,并选择打分最好的理论谱图对应的肽段作为该实验谱图的鉴定结果。整个匹配流程如下图:
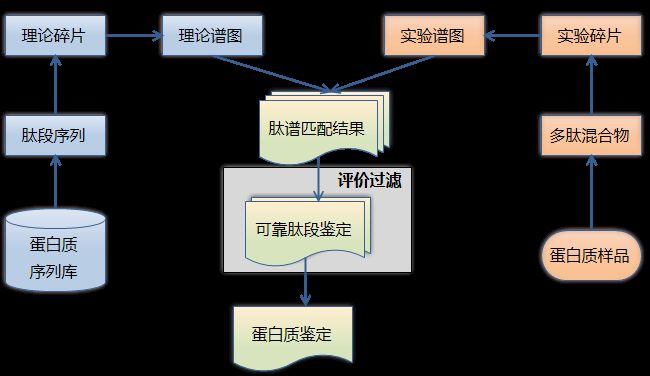
蛋白鉴定流程(版权所有:中科院计算所)
如果我告诉你,通过这个流程匹配到的肽段,并不一定都是正确的,你一定很想知道这其中的缘由吧!
概括来说,造成错误匹配的原因主要有以下几点:
1:蛋白质序列库不完整或者存在测序错误;
2:未知修饰,导致谱图难以被正确鉴定;
3:酶切实验的偏差,比如错切、漏切等;
4:母离子或子离子质量偏差;
5:搜索引擎的打分无法区分谱图对应的正确肽段和错误肽段,因此需要评价肽段鉴定的可靠性,搜索引擎才能根据鉴定到的可信肽段序列进行蛋白质推断。
看了上面的若干影响因素,大家是不是觉得,谱图匹配的正确性貌似很难判断啊?事实上,也的确很困难,但办法总比困难多,针对搜索引擎的打分如何评价肽段鉴定的可靠性这点上,作为行走江湖之必备神器--统计学就派上用场了!

说到评价肽段鉴定可靠性的常用统计指标,比如p值、E值、假发现率(False Discovery Rate, FDR)、q值和后验错误率(Posterior Error Probability, PEP),小编先抛出一个总表,你要挺住,别被砸晕喽~

评价肽段鉴定可靠性的统计指标(版权所有:中科院计算所)
可能你会说,我要是能看得懂这些公式,我还需要点开你这篇推文学习么?讲真,生物背景的童鞋们对数学公式的恐惧,小编十分理解和同情!所以接下来,让这些公式都乘凉去吧,我们用博大精深的汉语来聊一聊这些神奇的数学符号都是些什么玩意儿~
p-value
首先,大家见得最多的当然是p-value了!大家摸着良心想一想,你难道从来没有一次哪怕在心里默默地问过:"p-value小于0.05"?到底是几个意思啊?

要简单理解p-value,我们得先来说几个专业术语:原假设(或零假设 null hypothesis)和备则假设(althernative hypothesis)。啥叫原假设呢?就是需要我们收集证据来反对的假设,而备则假设就是需要收集证据来支持的假设。所以,原假设和备则假设正好是严格对立的,我们如果需要证明备则假设是真的,那么,我们要有强有力的证据推翻原假设。
科学的证伪精神,你感受到了吗?
我们还是举个例子先~ 例如你有一枚硬币,你想通过抛硬币的方式来测试它的正反面是不是均匀的。

首先,你写好原假设与备则假设,如下:
原假设:硬币是均匀的
备则假设:硬币不是均匀的
然后,你拿这枚硬币抛了20次,结果有14次是正面,有6次是反面。这个结果能否够强,可以推翻原假设呢?这时候我们就需要先算一下它的p-value。
咋算呢?就是把抛了20次,得到大于等于14次正面的所有情况的概率加起来,也就是:
得到14次正面的概率+得到15次正面的概率+ ...... +得到20次正面的概率
公式如下,不喜欢看公式的可以直接忽略:
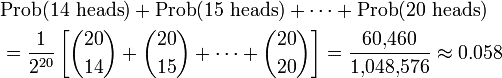
你只需要知道结论就是,这次试验的p-value等于0.058,如果我们要求p-value小于0.05,则该结果不够强到可以推翻“硬币是均匀的”这个原假设,但如果我们要求p-value小于0.06,可以推翻原假设了。
感受到了吗?这就是设p-value阈值的重要性!以我们的日常经验,抛一枚均匀硬币20次,得到14次正面(或反面)的可能性还是比较大的,如果以此就认为硬币是不均匀的,很难让人信服。
但如果我们抛了20次,得到了19次正面(或反面),或者我们抛了200次,得到180次正面(或反面),这样的结果如果拿来证明硬币是不均匀的,说服力是不是就强很多了呢?
那么,回到蛋白鉴定的问题上,同样地,我们先写好假设:
原假设:这个谱图匹配是错误的
备则假设:这个谱图匹配是正确的
如果我们能收集到足够有力的证据来反对这是错误匹配的假设,支持这是正确匹配的假设,我们就可以说,这个匹配是正确匹配的统计显著性就很高了!
换句话说,当p-value越大,则反对原假设的证据就越弱,反之,p-value越小,则反对原假设的证据就越强。具体到肽谱匹配的评价问题上,设某张谱图对应的肽谱匹配打分为x,则这个肽谱匹配的p-value指的是该谱图对应的随机匹配(即)打分大于或等于x的概率(对应抛硬币出现正面的次数大于等于14次的概率)。
如果随机匹配打分大于或等于x的概率越小的,即一个肽谱匹配的p值就越小,表明该匹配不是错误匹配的显著性就越大!
事实上,谱图匹配的时候,计算p-value要比抛硬币实验复杂很多,所以我们常常用近似的算法进行拟合,这种算法具体是怎样的,感兴趣的童鞋可以查阅类似如下的一些参考文献。
图片来源:PubMed
那么,p-value设多少才够小呢?这个问题其实很难回答,因为它并没有一个固定的答案,需要具体问题具体分析(虽然大家通常都很烦这句话...)。下面"E-value"的章节我们先举个例子,大家感受一下。
E-value
在常规的蛋白质序列库搜索中,一张谱图往往有多条候选肽,到底选哪一条?我们就需要评价某张谱图对应的打分最好的肽谱匹配的可靠性。
举个比较极端的例子:如果某一张谱图有500条候选肽,并且它们都不是谱图对应的真实答案。假设这500个肽谱匹配中的至少存在一个匹配打分的p值小于等于0.01,则这种匹配出现的概率为 1-(1-0.01)500≈0.99
500个随机匹配中打分最好的匹配的p值小于等于0.01的概率接近于1!也就是说,p值小于等于0.01的筛选也完全无法避免错误匹配的混入啊!难道,这么严格的取值都不足以作为这个肽谱匹配是正确的证据了?!

很遗憾地告诉你,当候选肽段很多的时候,这个证据确实还不够强!
那么,这种情况下,我们应该继续减小p-value的阈值吗?非也!因为这个问题不是出在p-value阈值定高定低了,而是出在p-value指标体现不出该谱图对应的候选肽段的数目。所以我们应该做的是,引入另一个既考虑了p-value又考虑了候选肽数目的统计指标,它就是传说中的E-value!它的别名就是:期望值~
老规矩,掉个书袋先,期望值的数学定义是:在一个离散性随机变量试验中每次可能结果的概率乘以其结果的总和。
晕了没?其实放到谱图匹配中,E-value的计算很简单,假设某张谱图有n条候选肽,它等于p-value乘以n:
E-value = p-value*n
这么简洁的公式,应该不会觉得可怕了吧?事实上,目前比较知名的一些搜索引擎都使用了E值,比如pFind[3]、Mascot[4]以及X!Tandem[5]等。
是不是大家现在一脸懵圈?这就对了。如果现在碰到和你一样懵或更懵的人的概率是万分之一,对面有1万个人,那么,懵圈的E-value就是1(别问我怎么算的)。也就是说,对面大概有1个人和你一样懵或更懵。
对一张二级谱图来说,鉴定软件报告一个肽段A与其匹配的E-value是1,就意味着其他候选肽中还有一个肽段B(虽然不知道B一定是谁)也可以与这个谱图匹配得不错,至少和肽段A与该谱图的匹配程度相当或更好。那么,软件报告这张谱图匹配肽段A就极有可能是假阳性结果。我们肯定是希望,E-value越小越好,E-value为0.001,就是只有千分之一个肽段可以威胁当前的匹配(突然想到电影院里半个人都没有的梗)。Mascot中常用 的阈值30分就是这么来的:
30分 = -10*log10(E-value=0.001)
你懂了么?
今天聊了p-value和E-value,不知道大家看下来感受如何?讲真,小编觉得能全篇读完的童鞋,也是不容易的!如果有啥疑惑,尽管给我们留言吧,小编争取做到有问必答!
下一篇我们会接着聊FDR和q-value,还能挺住的话,就请期待吧~
参考文献
[1] Ye D, Fu Y, Sun R X, et al. Open MS/MS spectral library search to identifyunanticipated post-translational modifications and increase spectralidentification rate. Bioinformatics, 2010, 26(12):i399-i40
[2] Lam H, Deutsch E W, Eddes J S, et al. Development and validation of aspectral library searching method for peptide identification from MS/MS.Proteomics, 2007, 7(5): 655-67
[3] Li D Q, Fu Y, Sun R X, et al. pFind: a noveldatabase-searching software system for automated peptide and proteinidentification via tandem mass spectrometry. Bioinformatics, 2005, 21(13): 3049-50
[4] Perkins D N, Pappin D J C, Creasy D M, et al. Probability-based proteinidentification by searching sequence databases using mass spectrometry data.Electrophoresis, 1999, 20(18): 3551-67
[5] Craig R, Beavis R C. TANDEM: matching proteins with tandem mass spectra.Bioinformatics, 2004, 20(9): 1466-7