
简介
在本教程中,我们将对PBMC的两个scATAC-seq数据集(5K和10K)和一个scRNA-seq数据集进行整合分析。这三个数据集均来自10X genomics测序平台产生的数据,可以直接在10x官网下载使用。
具体来说,我们将主要执行以下分析内容:
- Cell selection for PBMC 5k and 10k scATAC;
- Randomly sample 10,000 cells as landmarks;
- Unsupervised clustering of landmarks;
- Project the remaining (query) cells onto the landmarks;
- Supervised annotation of scATAC clusters using scRNA dataset;
- Downstream analysis including peak calling, differential analysis, prediction of gene-enhancer pairing.
分析步骤
- Step 0. Data download
- Step 1. Barcode selection
- Step 2. Add cell-by-bin matrix
- Step 3. Matrix binarization
- Step 4. Bin filtering
- Step 5. Dimensionality reduction of landmarks
- Step 6. Determine significant components
- Step 7. Graph-based clustering
- Step 8. Visualization
- Step 9. scRNA-seq based annotation
- Step 10. Create psudo multiomics cells
- Step 11. Remove cells of low prediction score
- Step 12. Gene expression projected into UMAP
- Step 13. Identify peak
- Step 14. Create a cell-by-peak matrix
- Step 15. Identify differentially accessible regions
- Step 16. Motif variability analysis
- Step 17. De novo motif discovery
- Step 18. Predict gene-enhancer pairs
Step 0. Data Download
开始分析之前,我们需要下载snaptools生成的snap文件和cellranger-atac产生的singlecell.csv文件。
# 下载所需的数据集和基因注释信息
$ wget http://renlab.sdsc.edu/r3fang//share/github/PBMC_ATAC_RNA/atac_pbmc_5k_nextgem.snap
$ wget http://cf.10xgenomics.com/samples/cell-atac/1.1.0/atac_pbmc_5k_nextgem/atac_pbmc_5k_nextgem_singlecell.csv
$ wget http://renlab.sdsc.edu/r3fang//share/github/PBMC_ATAC_RNA/atac_pbmc_10k_nextgem.snap
$ wget http://cf.10xgenomics.com/samples/cell-atac/1.1.0/atac_pbmc_10k_nextgem/atac_pbmc_10k_nextgem_singlecell.csv
$ wget http://cf.10xgenomics.com/samples/cell-atac/1.1.0/atac_pbmc_10k_nextgem/hg19.blacklist.bed.gz
$ wget http://cf.10xgenomics.com/samples/cell-atac/1.1.0/gencode.v19.annotation.gene.bed
Step 1. Barcode selection
首先,我们根据以下两个主要的标准来选择高质量的barcodes:
- number of unique fragments;
- fragments in promoter ratio;
# 加载所需R包
> library(SnapATAC);
> snap.files = c(
"atac_pbmc_5k_nextgem.snap",
"atac_pbmc_10k_nextgem.snap"
);
> sample.names = c(
"PBMC 5K",
"PBMC 10K"
);
> barcode.files = c(
"atac_pbmc_5k_nextgem_singlecell.csv",
"atac_pbmc_10k_nextgem_singlecell.csv"
);
# 读取snap文件
> x.sp.ls = lapply(seq(snap.files), function(i){
createSnap(
file=snap.files[i],
sample=sample.names[i]
);
})
> names(x.sp.ls) = sample.names;
# 读取barcode信息
> barcode.ls = lapply(seq(snap.files), function(i){
barcodes = read.csv(
barcode.files[i],
head=TRUE
);
# remove NO BAROCDE line
barcodes = barcodes[2:nrow(barcodes),];
barcodes$logUMI = log10(barcodes$passed_filters + 1);
barcodes$promoter_ratio = (barcodes$promoter_region_fragments+1) / (barcodes$passed_filters + 1);
barcodes
})
# 质控指标数据可视化
> plots = lapply(seq(snap.files), function(i){
p1 = ggplot(
barcode.ls[[i]],
aes(x=logUMI, y=promoter_ratio)) +
geom_point(size=0.3, col="grey") +
theme_classic() +
ggtitle(sample.names[[i]]) +
ylim(0, 1) + xlim(0, 6) +
labs(x = "log10(UMI)", y="promoter ratio")
p1
})
> plots

# 查看snap对象信息
> x.sp.ls
## $`PBMC 5K`
## number of barcodes: 20000
## number of bins: 0
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
##
## $`PBMC 10K`
## number of barcodes: 20000
## number of bins: 0
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
# for both datasets, we identify usable barcodes using [3.5-5] for log10(UMI) and [0.4-0.8] for promoter ratio as cutoff.
# 设定质控阈值进行筛选过滤
> cutoff.logUMI.low = c(3.5, 3.5);
> cutoff.logUMI.high = c(5, 5);
> cutoff.FRIP.low = c(0.4, 0.4);
> cutoff.FRIP.high = c(0.8, 0.8);
# 根据过滤的阈值进行barcode的筛选
> barcode.ls = lapply(seq(snap.files), function(i){
barcodes = barcode.ls[[i]];
idx = which(
barcodes$logUMI >= cutoff.logUMI.low[i] &
barcodes$logUMI <= cutoff.logUMI.high[i] &
barcodes$promoter_ratio >= cutoff.FRIP.low[i] &
barcodes$promoter_ratio <= cutoff.FRIP.high[i]
);
barcodes[idx,]
});
> x.sp.ls = lapply(seq(snap.files), function(i){
barcodes = barcode.ls[[i]];
x.sp = x.sp.ls[[i]];
barcode.shared = intersect(x.sp@barcode, barcodes$barcode);
x.sp = x.sp[match(barcode.shared, x.sp@barcode),];
barcodes = barcodes[match(barcode.shared, barcodes$barcode),];
x.sp@metaData = barcodes;
x.sp
})
> names(x.sp.ls) = sample.names;
> x.sp.ls
## $`PBMC 5K`
## number of barcodes: 4526
## number of bins: 0
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
##
## $`PBMC 10K`
## number of barcodes: 9039
## number of bins: 0
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
# combine two snap object
# combine two snap object
# 使用Reduce函数将两个snap对象进行合并
> x.sp = Reduce(snapRbind, x.sp.ls);
> x.sp@metaData["sample"] = x.sp@sample;
> x.sp
## number of barcodes: 13565
## number of bins: 0
## number of genes: 0
## number of peaks: 0
> table(x.sp@sample);
## PBMC 10K PBMC 5K
## 9039 4526
Step 2. Add cell-by-bin matrix
接下来,我们使用addBmatToSnap函数生成5kb分辨率的cell-by-bin计数矩阵,并将其添加到snap对象中。该函数将自动从两个snap文件中读取cell-by-bin矩阵,并将连接后的矩阵添加到snap对象的bmat属性中。
# 使用addBmatToSnap函数计算cell-by-bin计数矩阵
> x.sp = addBmatToSnap(x.sp, bin.size=5000);
Step 3. Matrix binarization
我们使用makeBinary函数将cell-by-bin计数矩阵转换为二进制矩阵。在count矩阵中,某些项具有异常高的覆盖率,这可能是由比对错误造成的。因此,我们将删除计数矩阵中top 0.1%的项,并将其余非零的值转换为1。
# 使用makeBinary函数将cell-by-bin计数矩阵转换成二进制矩阵
> x.sp = makeBinary(x.sp, mat="bmat");
Step 4. Bin filtering
首先,我们过滤掉任何与ENCODE中blacklist区域重叠的bins,以防止潜在的artifacts。
> library(GenomicRanges);
# 读取blacklist信息
> black_list = read.table("hg19.blacklist.bed.gz");
> black_list.gr = GRanges(
black_list[,1],
IRanges(black_list[,2], black_list[,3])
);
> idy = queryHits(
findOverlaps(x.sp@feature, black_list.gr)
);
> if(length(idy) > 0){
x.sp = x.sp[,-idy, mat="bmat"];
};
> x.sp
## number of barcodes: 13565
## number of bins: 624794
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
接下来,我们移除那些不需要的染色体信息。
> chr.exclude = seqlevels(x.sp@feature)[grep("random|chrM", seqlevels(x.sp@feature))];
> idy = grep(paste(chr.exclude, collapse="|"), x.sp@feature);
> if(length(idy) > 0){
x.sp = x.sp[,-idy, mat="bmat"]
};
> x.sp
## number of barcodes: 13565
## number of bins: 624297
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
第三,bins的覆盖率大致是服从对数正态分布的。我们将与不变特征(如管家基因的启动子)重叠的前5%的bins进行删除 。
> bin.cov = log10(Matrix::colSums(x.sp@bmat)+1);
> hist(
bin.cov[bin.cov > 0],
xlab="log10(bin cov)",
main="log10(Bin Cov)",
col="lightblue",
xlim=c(0, 5)
);
> bin.cutoff = quantile(bin.cov[bin.cov > 0], 0.95);
> idy = which(bin.cov <= bin.cutoff & bin.cov > 0);
> x.sp = x.sp[, idy, mat="bmat"];
> x.sp
## number of barcodes: 13565
## number of bins: 534985
## number of genes: 0
## number of peaks: 0
## number of motifs: 0

最后,我们将进一步删除bin覆盖率小于1000的任何细胞,这是因为尽管有些细胞可能含有较高的unique fragments片段,但是过滤后的bin覆盖率却很低。此步骤是可选的,但强烈建议这样做。
> idx = which(Matrix::rowSums(x.sp@bmat) > 1000);
> x.sp = x.sp[idx,];
> x.sp
## number of barcodes: 13434
## number of bins: 534985
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
Step 5. Dimentionality reduction
SnapATAC采用diffusion maps算法进行数据降维,这是一种非线性降维的技术,它通过对数据执行random walk来发现低维的manifold,并且对噪音和扰动具有很强的鲁棒性。
但是,diffusion maps算法的计算成本会随着细胞数目的增加而呈现指数级增长的趋势。为了克服这一局限,我们将Nyström方法(a sampling technique)和diffusion maps算法相结合,给出Nyström Landmark diffusion map来生成大规模数据集的低维嵌入。
Nyström landmark diffusion maps算法主要包括以下三个步骤:
-
sampling: sample a subset of K (K≪N) cells from N total cells as “landmarks”. Instead of random sampling, here we adopted a density-based sampling approach developed in SCTransform to preserve the density distribution of the N original points; -
embedding: compute a diffusion map embedding for K landmarks; -
extension: project the remaining N-K cells onto the low-dimensional embedding as learned from the landmarks to create a joint embedding space for all cells.
在本示例中,我们将采样10,000个细胞作为landmarks,并将余下的query细胞投射到嵌入landmarks的diffusion maps上。
> row.covs.dens <- density(
x = x.sp@metaData[,"logUMI"],
bw = 'nrd', adjust = 1
);
> sampling_prob <- 1 / (approx(x = row.covs.dens$x, y = row.covs.dens$y, xout = x.sp@metaData[,"logUMI"])$y + .Machine$double.eps);
> set.seed(1);
> idx.landmark.ds <- sort(sample(x = seq(nrow(x.sp)), size = 10000, prob = sampling_prob));
将x.sp对象分割为landmark(x.landmark.sp)细胞和query(x.query.sp)细胞。
> x.landmark.sp = x.sp[idx.landmark.ds,];
> x.query.sp = x.sp[-idx.landmark.ds,];
Run diffusion maps on the landmark cells.
使用landmark细胞进行diffusion maps降维处理
> x.landmark.sp = runDiffusionMaps(
obj= x.landmark.sp,
input.mat="bmat",
num.eigs=50
);
> x.landmark.sp@metaData$landmark = 1;
Porject query cells to landmarks.
将query细胞映射到landmarks上
> x.query.sp = runDiffusionMapsExtension(
obj1=x.landmark.sp,
obj2=x.query.sp,
input.mat="bmat"
);
> x.query.sp@metaData$landmark = 0;
Combine landmark and query cells.
合并landmark和query细胞
Note: To merge snap objects, all the matrix (bmat, gmat, pmat) and metaData must be of the same number of columns between snap objects.
> x.sp = snapRbind(x.landmark.sp, x.query.sp);
> x.sp = x.sp[order(x.sp@metaData[,"sample"])]; #IMPORTANT
Step 6. Determine significant components
接下来,我们将确定特征向量(eigen-vectors)的数目,以便用于后续的分析。在下面的例子中,我们选择前15个特征向量。
> plotDimReductPW(
obj=x.sp,
eigs.dims=1:50,
point.size=0.3,
point.color="grey",
point.shape=19,
point.alpha=0.6,
down.sample=5000,
pdf.file.name=NULL,
pdf.height=7,
pdf.width=7
);

Step 7. Graph-based clustering
接下来,我们使用所选的特征向量成分,来构造K近邻(KNN)聚类图。其中,每个节点是一个细胞,根据欧氏距离来确定每个细胞的k个近邻细胞。
> x.sp = runKNN(
obj=x.sp,
eigs.dims=1:20,
k=15
);
> library(leiden);
> x.sp=runCluster(
obj=x.sp,
tmp.folder=tempdir(),
louvain.lib="leiden",
seed.use=10,
resolution=0.7
);
Step 8. Visualization
SnapATAC可以使用tSNE(FI-tsne)和UMAP方法对降维聚类后的细胞进行可视化的展示和探索。在本示例中,我们使用UMAP方法进行展示。
> library(umap);
> x.sp = runViz(
obj=x.sp,
tmp.folder=tempdir(),
dims=2,
eigs.dims=1:20,
method="umap",
seed.use=10
);
> par(mfrow = c(2, 2));
> plotViz(
obj= x.sp,
method="umap",
main="Cluster",
point.color=x.sp@cluster,
point.size=0.2,
point.shape=19,
text.add=TRUE,
text.size=1,
text.color="black",
down.sample=10000,
legend.add=FALSE
);
> plotFeatureSingle(
obj=x.sp,
feature.value=x.sp@metaData[,"logUMI"],
method="umap",
main="Read Depth",
point.size=0.2,
point.shape=19,
down.sample=10000,
quantiles=c(0.01, 0.99)
);
> plotViz(
obj= x.sp,
method="umap",
main="Sample",
point.size=0.2,
point.shape=19,
point.color=x.sp@sample,
text.add=FALSE,
text.size=1.5,
text.color="black",
down.sample=10000,
legend.add=TRUE
);
> plotViz(
obj= x.sp,
method="umap",
main="Landmark",
point.size=0.2,
point.shape=19,
point.color=x.sp@metaData[,"landmark"],
text.add=FALSE,
text.size=1.5,
text.color="black",
down.sample=10000,
legend.add=TRUE
);
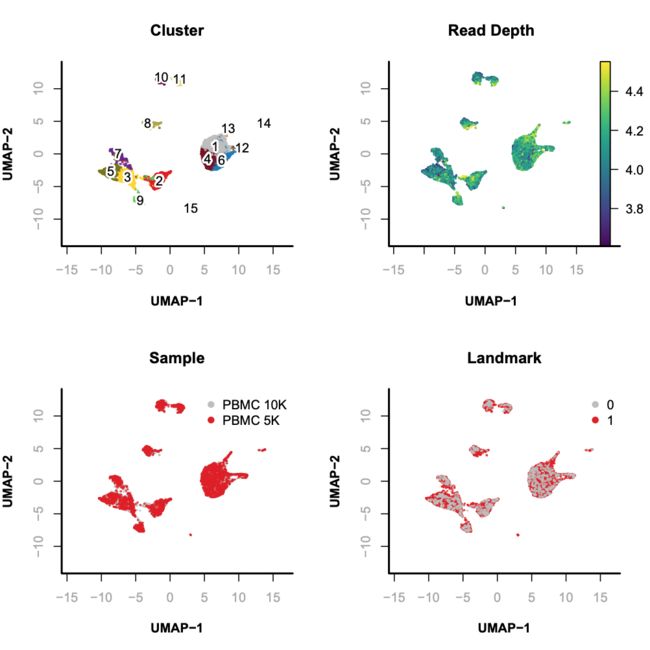
Step 9. scRNA-seq based annotation
在本示例中,我们将使用相应的scRNA-seq数据集来对scATAC-seq数据的细胞类群进行注释。我们可以通过(https://www.dropbox.com/s/3f3p5nxrn5b3y4y/pbmc_10k_v3.rds?dl=1)地址下载所需的10X PBMC scRNA-seq(pbmc_10k_v3.rds)的Seurat对象。
> library(Seurat);
> pbmc.rna = readRDS("pbmc_10k_v3.rds");
> pbmc.rna$tech = "rna";
> variable.genes = VariableFeatures(object = pbmc.rna);
> genes.df = read.table("gencode.v19.annotation.gene.bed");
> genes.gr = GRanges(genes.df[,1], IRanges(genes.df[,2], genes.df[,3]), name=genes.df[,4]);
> genes.sel.gr = genes.gr[which(genes.gr$name %in% variable.genes)];
## reload the bmat, this is optional but highly recommanded
> x.sp = addBmatToSnap(x.sp);
> x.sp = createGmatFromMat(
obj=x.sp,
input.mat="bmat",
genes=genes.sel.gr,
do.par=TRUE,
num.cores=10
);
接下来,我们将snap对象转换为Seurat对象,用于后续的数据整合。
# 使用snapToSeurat函数将snap对象转换为Seurat对象
> pbmc.atac <- snapToSeurat(
obj=x.sp,
eigs.dims=1:20,
norm=TRUE,
scale=TRUE
);
# 识别整合的Anchors信息
> transfer.anchors <- FindTransferAnchors(
reference = pbmc.rna,
query = pbmc.atac,
features = variable.genes,
reference.assay = "RNA",
query.assay = "ACTIVITY",
reduction = "cca"
);
# 使用TransferData函数进行数据映射整合
> celltype.predictions <- TransferData(
anchorset = transfer.anchors,
refdata = pbmc.rna$celltype,
weight.reduction = pbmc.atac[["SnapATAC"]],
dims = 1:20
);
> x.sp@metaData$predicted.id = celltype.predictions$predicted.id;
> x.sp@metaData$predict.max.score = apply(celltype.predictions[,-1], 1, max);
> x.sp@cluster = as.factor(x.sp@metaData$predicted.id);
Step 10. Create psudo multiomics cells
现在,snap对象x.sp中包含了的每个细胞的染色质可及性@bmat和基因表达@gmat的信息。
> refdata <- GetAssayData(
object = pbmc.rna,
assay = "RNA",
slot = "data"
);
> imputation <- TransferData(
anchorset = transfer.anchors,
refdata = refdata,
weight.reduction = pbmc.atac[["SnapATAC"]],
dims = 1:20
);
> x.sp@gmat = t(imputation@data);
> rm(imputation); # free memory
> rm(refdata); # free memory
> rm(pbmc.rna); # free memory
> rm(pbmc.atac); # free memory
Step 11. Remove cells of low prediction score
> hist(
x.sp@metaData$predict.max.score,
xlab="prediction score",
col="lightblue",
xlim=c(0, 1),
main="PBMC 10X"
);
> abline(v=0.5, col="red", lwd=2, lty=2);
> table(x.sp@metaData$predict.max.score > 0.5);
## FALSE TRUE
## 331 13103
> x.sp = x.sp[x.sp@metaData$predict.max.score > 0.5,];
> x.sp
## number of barcodes: 13045
## number of bins: 627478
## number of genes: 19089
## number of peaks: 0
> plotViz(
obj=x.sp,
method="umap",
main="PBMC 10X",
point.color=x.sp@metaData[,"predicted.id"],
point.size=0.5,
point.shape=19,
text.add=TRUE,
text.size=1,
text.color="black",
down.sample=10000,
legend.add=FALSE
);

Step 12. Gene expression projected onto UMAP
接下来,我们使用UMAP方法对一些marker基因的表达水平进行可视化展示。
> marker.genes = c(
"IL32", "LTB", "CD3D",
"IL7R", "LDHB", "FCGR3A",
"CD68", "MS4A1", "GNLY",
"CD3E", "CD14", "CD14",
"FCGR3A", "LYZ", "PPBP",
"CD8A", "PPBP", "CST3",
"NKG7", "MS4A7", "MS4A1",
"CD8A"
);
> par(mfrow = c(3, 3));
> for(i in 1:9){
j = which(colnames(x.sp@gmat) == marker.genes[i])
plotFeatureSingle(
obj=x.sp,
feature.value=x.sp@gmat[,j],
method="umap",
main=marker.genes[i],
point.size=0.1,
point.shape=19,
down.sample=10000,
quantiles=c(0.01, 0.99)
)};

Step 13. Identify peaks
接下来,我们将每个cluster群中的reads进行聚合,创建用于peak calling和可视化的集成track。执行完peak calling后,将生成一个.narrowPeak的文件,该文件中包含了识别出的peaks信息,和一个.bedgraph的文件,可以用于可视化展示。为了获得最robust的结果,我们不建议对细胞数目小于100的cluster群执行此步骤。
> system("which snaptools");
/home/r3fang/anaconda2/bin/snaptools
> system("which macs2");
/home/r3fang/anaconda2/bin/macs2
# 使用runMACS函数调用macs2进行peak calling
> peaks = runMACS(
obj=x.sp[which(x.sp@cluster=="CD4 Naive"),],
output.prefix="PBMC.CD4_Naive",
path.to.snaptools="/home/r3fang/anaconda2/bin/snaptools",
path.to.macs="/home/r3fang/anaconda2/bin/macs2",
gsize="hs", # mm, hs, etc
buffer.size=500,
num.cores=10,
macs.options="--nomodel --shift 100 --ext 200 --qval 5e-2 -B --SPMR",
tmp.folder=tempdir()
);
为了对所有的cluster群执行peak calling,我们提供了一个简便的脚本来实现该步骤。
# call peaks for all cluster with more than 100 cells
# 选出那些细胞数目大于100的cluster群
> clusters.sel = names(table(x.sp@cluster))[which(table(x.sp@cluster) > 100)];
# 批量对不同的cluster进行peak calling
> peaks.ls = mclapply(seq(clusters.sel), function(i){
print(clusters.sel[i]);
peaks = runMACS(
obj=x.sp[which(x.sp@cluster==clusters.sel[i]),],
output.prefix=paste0("PBMC.", gsub(" ", "_", clusters.sel)[i]),
path.to.snaptools="/home/r3fang/anaconda2/bin/snaptools",
path.to.macs="/home/r3fang/anaconda2/bin/macs2",
gsize="hs", # mm, hs, etc
buffer.size=500,
num.cores=1,
macs.options="--nomodel --shift 100 --ext 200 --qval 5e-2 -B --SPMR",
tmp.folder=tempdir()
);
peaks
}, mc.cores=5);
> peaks.names = system("ls | grep narrowPeak", intern=TRUE);
> peak.gr.ls = lapply(peaks.names, function(x){
peak.df = read.table(x)
GRanges(peak.df[,1], IRanges(peak.df[,2], peak.df[,3]))
})
# 合并peak信息
> peak.gr = reduce(Reduce(c, peak.gr.ls));
我们可以将每个cluster群生成的bdg文件导入到IGV或其他基因组浏览器(如UW genome browser)进行可视化的展示,下面是来自UW genome browser的FOXJ2基因及其侧翼区域的screenshot。

Step 14. Create a cell-by-peak matrix
接下来,我们使用合并后的peaks作为参考,来创建一个cell-by-peak计数矩阵,并将其添加到snap对象中。
首先,我们将合并后的peaks信息写入到peaks.combined.bed文件中
> peaks.df = as.data.frame(peak.gr)[,1:3];
> write.table(peaks.df,file = "peaks.combined.bed",append=FALSE,
quote= FALSE,sep="\t", eol = "\n", na = "NA", dec = ".",
row.names = FALSE, col.names = FALSE, qmethod = c("escape", "double"),
fileEncoding = "")
接下来,我们创建一个cell-by-peak计数矩阵,并将其添加到snap对象中。这一步可能需要一段时间。
$ snaptools snap-add-pmat \
--snap-file atac_pbmc_10k_nextgem.snap \
--peak-file peaks.combined.bed &
$ snaptools snap-add-pmat \
--snap-file atac_pbmc_5k_nextgem.snap \
--peak-file peaks.combined.bed
然后,我们将计算好的cell-by-peak计数矩阵添加到已有的snap对象中。
> x.sp = addPmatToSnap(x.sp);
Step 15. Identify differentially accessible peaks
对于一组给定的细胞Ci,我们首先在diffusion component空间中寻找邻近的细胞Cj (|Ci|=|Cj|)作为“background”细胞进行比较。如果Ci细胞的数目占总细胞数目的一半以上,则使用剩余的细胞作为local background。接下来,我们将Ci和Cj细胞进行聚合,来创建两个raw-count向量,即Vci和Vcj。然后,我们使用R包edgeR (v3.18.1)的精确检验(exact test)来对Vci和Vcj进行差异分析,其中,对于小鼠设置BCV=0.1,而人类设置BCV= 0.4。最后,再使用Benjamini-Hochberg多重检验校正方法,来调整p-value值为错误发现率(FDR)值。对于FDR值小于0.05的peaks,被选为显著的DARs。
我们发现这种方法也存在一定的局限性,特别是对于那些细胞数目较少的cluster群,统计的有效性可能不够强大。对于那些未能识别出显著差异peaks的cluster群,我们将根据富集p-value值对元素进行排序,并挑选出最具代表性的2,000个peaks进行后续的motif分析。
> DARs = findDAR(
obj=x.sp,
input.mat="pmat",
cluster.pos="CD14+ Monocytes",
cluster.neg.method="knn",
test.method="exactTest",
bcv=0.4, #0.4 for human, 0.1 for mouse
seed.use=10
);
> DARs$FDR = p.adjust(DARs$PValue, method="BH");
> idy = which(DARs$FDR < 5e-2 & DARs$logFC > 0);
> par(mfrow = c(1, 2));
> plot(DARs$logCPM, DARs$logFC,
pch=19, cex=0.1, col="grey",
ylab="logFC", xlab="logCPM",
main="CD14+ Monocytes"
);
> points(DARs$logCPM[idy],
DARs$logFC[idy],
pch=19,
cex=0.5,
col="red"
);
> abline(h = 0, lwd=1, lty=2);
> covs = Matrix::rowSums(x.sp@pmat);
> vals = Matrix::rowSums(x.sp@pmat[,idy]) / covs;
> vals.zscore = (vals - mean(vals)) / sd(vals);
> plotFeatureSingle(
obj=x.sp,
feature.value=vals.zscore,
method="umap",
main="CD14+ Monocytes",
point.size=0.1,
point.shape=19,
down.sample=5000,
quantiles=c(0.01, 0.99)
);
Step 16. Motif variability analysis
SnapATAC可以调用chromVAR(Schep et al)程序进行motif可变性分析。
# 加载所需的R包
> library(chromVAR);
> library(motifmatchr);
> library(SummarizedExperiment);
> library(BSgenome.Hsapiens.UCSC.hg19);
> x.sp = makeBinary(x.sp, "pmat");
# 使用runChromVAR函数调用ChromVAR进行motif可变性分析
> x.sp@mmat = runChromVAR(
obj=x.sp,
input.mat="pmat",
genome=BSgenome.Hsapiens.UCSC.hg19,
min.count=10,
species="Homo sapiens"
);
> x.sp;
## number of barcodes: 13103
## number of bins: 627478
## number of genes: 19089
## number of peaks: 157750
## number of motifs: 271
> motif_i = "MA0071.1_RORA";
> dat = data.frame(x=x.sp@metaData$predicted.id, y=x.sp@mmat[,motif_i]);
> p <- ggplot(dat, aes(x=x, y=y, fill=x)) +
theme_classic() +
geom_violin() +
xlab("cluster") +
ylab("motif enrichment") +
ggtitle("MA0071.1_RORA") +
theme(
plot.margin = margin(5,1,5,1, "cm"),
axis.text.x = element_text(angle = 90, hjust = 1),
axis.ticks.x=element_blank(),
legend.position = "none"
);

Step 17. De novo motif discovery
SnapATAC还可以调用homer用于对识别出的差异可及性区域(DARs)进行motif的识别和富集分析.
# 查看findMotifsGenome.pl程序安装的路径
> system("which findMotifsGenome.pl");
/projects/ps-renlab/r3fang/public_html/softwares/homer/bin/findMotifsGenome.pl
# 使用findDAR函数鉴定DAR区域
> DARs = findDAR(
obj=x.sp,
input.mat="pmat",
cluster.pos="Double negative T cell",
cluster.neg.method="knn",
test.method="exactTest",
bcv=0.4, #0.4 for human, 0.1 for mouse
seed.use=10
);
> DARs$FDR = p.adjust(DARs$PValue, method="BH");
> idy = which(DARs$FDR < 5e-2 & DARs$logFC > 0);
# 使用runHomer函数调用homer进行de novo motif discovery
> motifs = runHomer(
x.sp[,idy,"pmat"],
mat = "pmat",
path.to.homer = "/projects/ps-renlab/r3fang/public_html/softwares/homer/bin/findMotifsGenome.pl",
result.dir = "./homer/DoubleNegativeTcell",
num.cores=5,
genome = 'hg19',
motif.length = 10,
scan.size = 300,
optimize.count = 2,
background = 'automatic',
local.background = FALSE,
only.known = TRUE,
only.denovo = FALSE,
fdr.num = 5,
cache = 100,
overwrite = TRUE,
keep.minimal = FALSE
);

Step 18. Predict gene-enhancer pairs
最后,我们还可以使用一种“pseudo”细胞的方法,根据单个细胞中基因的表达和远端调控元件的染色质可及性的关系,将远端调控元件连接到目标基因上。对于给定的marker基因,我们首先识别出marker基因侧翼区域内的peak。对于每个侧翼的peak,我们使用基因表达作为输入变量进行逻辑回归来预测binarized的染色质状态。产生的结果估计了染色质可及性与基因表达之间联系的重要性。
> TSS.loci = GRanges("chr12", IRanges(8219067, 8219068));
> pairs = predictGenePeakPair(
x.sp,
input.mat="pmat",
gene.name="C3AR1",
gene.loci=resize(TSS.loci, width=500000, fix="center"),
do.par=FALSE
);
# convert the pair to genome browser arc plot format
> pairs.df = as.data.frame(pairs);
> pairs.df = data.frame(
chr1=pairs.df[,"seqnames"],
start1=pairs.df[,"start"],
end1=pairs.df[,"end"],
chr2="chr2",
start2=8219067,
end2=8219068,
Pval=pairs.df[,"logPval"]
);
> head(pairs.df)
## chr1 start1 end1 chr2 start2 end2 Pval
## 1 chr12 7984734 7985229 chr2 8219067 8219068 14.6075918
## 2 chr12 7987561 7988085 chr2 8219067 8219068 5.6718381
## 3 chr12 7989776 7990567 chr2 8219067 8219068 24.2564608
## 4 chr12 7996454 7996667 chr2 8219067 8219068 0.6411017
## 5 chr12 8000059 8000667 chr2 8219067 8219068 2.0324922
## 6 chr12 8012404 8013040 chr2 8219067 8219068 0.0000000

参考来源:https://gitee.com/booew/SnapATAC/blob/master/examples/10X_PBMC_15K/README.md