参考这篇
ID转换不用怕,clusterProfiler帮你忙
这篇详细讲了无参考基因组该怎么办,值得一看
简单介绍一下几种常用的ID:
Ensemble id:一般以ENSG开头,后边跟11位数字。如TP53基因:ENSG00000141510
Entrez id:通常为纯数字。如TP53基因:7157
Symbol id:为我们常在文献中报道的基因名称。如TP53基因的symbol id为TP53
Refseq id:NCBI提供的参考序列数据库:可以是NG、NM、NP开头,代表基因,转录本和蛋白质。如TP53基因的某个转录本信息可为NM_000546
1 转换ID
1.1 载入包library(clusterProfiler)
library(clusterProfiler)
library(DOSE)
library(org.Hs.eg.db)
org.Hs.eg.db是人类基因组注释,如果需要其他物种的注释信息,可以去bioconductor这里获得
1.2. keytypes()查看注释包中支持的ID转换类型,基本包括了所以常用的ID类型

1.3. bitr()函数转换ID
函数全部内容如下
bitr(geneID, fromType, toType, OrgDb, drop = TRUE
geneID:输入待转换的geneID
fromType:输入的ID类型
toType:希望输出的ID类型
OrgDb:注释对象的信息
drop:drop = FALSE 保留空值
以TP53为例,希望输出'ENTREZID','ENSEMBL','REFSEQ'
my_id <- c("TP53")
output <- bitr(my_id,
fromType = 'SYMBOL',
toType = c('ENTREZID','ENSEMBL','REFSEQ'),
OrgDb = 'org.Hs.eg.db')

1.4. bitr_kegg()函数进行基因ID与蛋白质ID的转换
函数全部内容如下,与bitr()类似
bitr_kegg(geneID, fromType, toType, organism, drop = TRUE)
在参数这里与
bitr()有些区别!
geneID:输入待转换的geneID
fromType:输入的ID类型只能是kegg(与entrez id相同),ncbi-geneid,ncbi-proteinidoruniprot中的一个
toType:希望输出的ID类型,也只能是kegg(与entrez id相同),ncbi-geneid,ncbi-proteinidoruniprot中的一个
organism:hsa,代表人类,其他的物种名称可以参考kegg的网站
drop:去除空值与否
TP53的entrez id为7157
kegg <- bitr_kegg(7157,
fromType = 'kegg',
toType = c('ncbi-geneid', 'ncbi-proteinid', 'uniprot'),
organism = 'hsa')
报错了,所以一次应该只能进行一种转换!

改成这样↓
kegg <- bitr_kegg(7157,
fromType = 'kegg',
toType = 'ncbi-proteinid',
organism = 'hsa')

转换为uniprot
kegg <- bitr_kegg(7157,
fromType = 'kegg',
toType = 'uniprot',
organism = 'hsa')

去UniProt网站上查询一下为什么会有三个:
P04637显示的是TP53基因的蛋白质表达水平,级别是Reviewed,就是其来源为UniProtKB/Swiss-Prot。

K7PPA8显示的是转录本水平的表达,级别是Unreviewed,就是其来源为UniProtKB/TrEMBL

Q53GA5显示的是转录本水平的表达,级别是Unreviewed,就是其来源为UniProtKB/TrEMBL

一般ID转换仅仅为开始的准备工作,将自己的数剧转换好之后可以进行后续的分析。
另外,利用clusterProfiler包可以进行许多丰富的下游分析,比如GO分析、KEGG分析等等
2 富集分析
参考这篇
最常用的基因注释工具是GO和KEGG注释,这基本上是和差异基因分析一样,必做的事,GO可在BP(生物过程),MF(分子功能),CC(细胞组分)三方面进行注释
 GO中的注释信息
GO中的注释信息
KEGG数据库全部的物种及其简写(三个字母)
有很多在线(DAVID、Gather、GOrilla,revigo)或者客户端版的工具(IPA(IPA不是用的GO和KEGG数据库)和FUNRICH)可以用来分析差异基因,我主要实践一下clusterProfiler
clusterProfiler提供有两种富集方式:
① ORA(Over-Representation Analysis)差异基因富集分析(不需要表达值,只需要gene name)
② FCS,它的代表就是GSEA,即基因集(gene set)富集分析(不管有无差异,需要全部genes表达值)
① ORA 差异基因富集分析(不需要表达值,只需要gene name)
1) 先读进来差异基因
sig.gene <- read.delim("你的路径/final_result.txt(之前计算出的差异基因文件)", sep = '\t', stringsAsFactors = FALSE, check.names = FALSE)
gene<-sig.gene[,1]
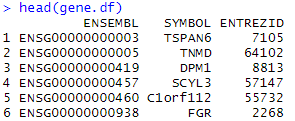
head(gene)
#转换ID
gene.df<-bitr(gene, fromType = "ENSEMBL",
toType = c("SYMBOL","ENTREZID"),
OrgDb = org.Hs.eg.db,
drop = FALSE)
head(gene.df)
2) GO富集
ego_cc<-enrichGO(gene = gene.df$ENSEMBL,
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',
ont = "CC",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05)
ego_bp<-enrichGO(gene = gene.df$ENSEMBL,
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05)
参数意义:
keyType 指定基因ID的类型,默认为ENTREZID
OrgDb 指定该物种对应的org包的名字
ont 代表GO的3大类别,BP, CC, MF,也可是全部ALL
pAdjustMethod 指定多重假设检验矫正的方法,有“ holm”, “hochberg”, “hommel”, “bonferroni”, “BH”, “BY”, “fdr”, “none”中的一种
cufoff 指定对应的阈值
readable=TRUE 代表将基因ID转换为gene symbol。
barplot() #富集柱形图
dotplot() #富集气泡图
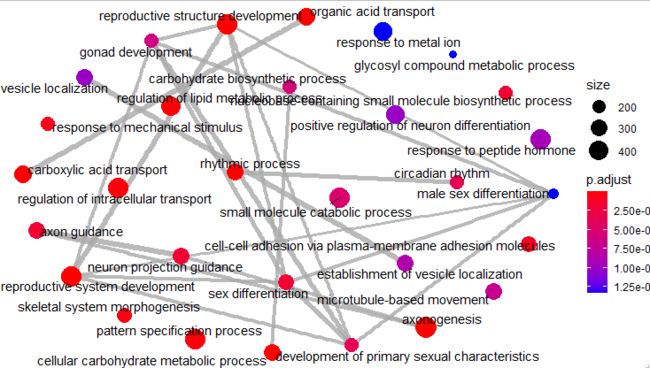
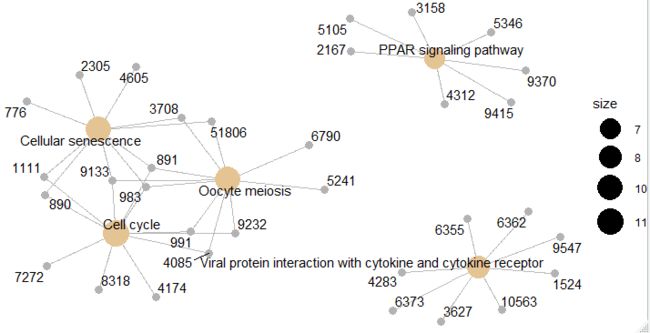
cnetplot() #网络图展示富集功能和基因的包含关系
emapplot() #网络图展示各富集功能之间共有基因关系
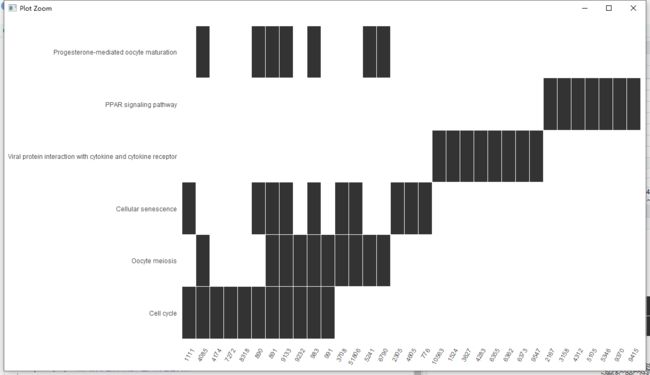
heatplot() #热图展示富集功能和基因的包含关系
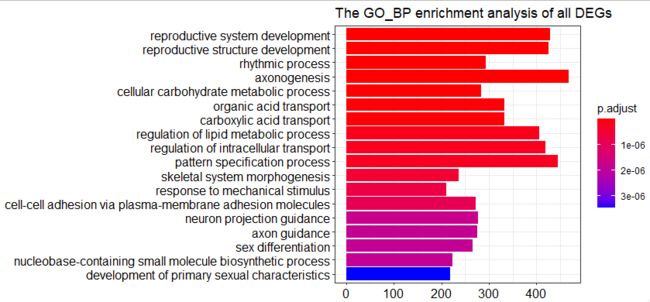
3) barplot()作图显示,依赖于ggplot2包
library(ggplot2)
barplot(ego_bp,showCategory = 18,title="The GO_BP enrichment analysis of all DEGs ")
ggplot2参数意义具体可以看这篇

4) 或者是KEGG分析
输入的gene要为vector格式的entrezid,可以用is.vector()判断是否是vector格式
library(stringr)
kk<-enrichKEGG(gene = gene.df$ENTREZID,
organism = "hsa",
keyType = "kegg",
pAdjustMethod = "BH",
pvalueCutoff = 0.01)

但是这样产生的
kk几乎都是Na,更别提画图了
于是我去看了Y叔的手册,发现需要使用
DOSE构建,改成
library(DOSE)
data(geneList, package = "DOSE")
gene <- names(geneList)[abs(geneList) > 2]
kk<-enrichKEGG(gene = gene,
organism = "hsa",
keyType = "kegg",
pAdjustMethod = "BH",
pvalueCutoff = 0.01)
就可以了,继续往下进行
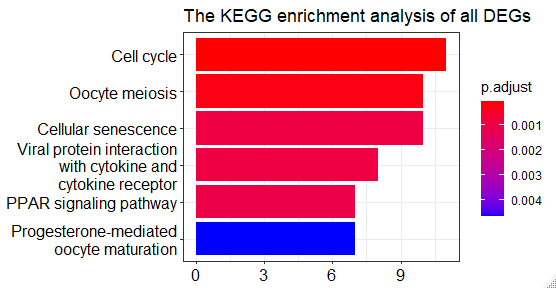
#bar图
barplot(kk,showCategory = 25, title="The KEGG enrichment analysis of all DEGs")+
scale_size(range=c(2, 12))+
scale_x_discrete(labels=function(kk) str_wrap(kk,width = 25))
#点图
dotplot(kk,showCategory = 25, title="The KEGG enrichment analysis of all DEGs")+
scale_size(range=c(2, 12))+
scale_x_discrete(labels=function(kk) str_wrap(kk,width = 25))

② GSEA 基因集(gene set)富集分析(不管有无差异,需要全部genes表达值)
这篇为网页版操作
好处:可以发现被差异基因舍弃的genes可能参与了某重要生理过程或信号通路
要点:用所有差异基因来跑GSEA,而不是按筛选条件筛选出来的,否则GSEA就失去了意义
1)读入全部差异基因数据
library(dplyr)
all_deseq <- read.delim("你的路径/DESeq2-res.txt", sep = '\t', stringsAsFactors = FALSE, check.names = FALSE)

2) 提取出gene_id 和 log2FoldChange列
ensemblid_log2fdc <-dplyr::select(all_deseq, gene_id, log2FoldChange)
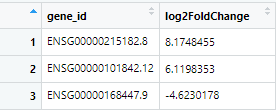
3) 可以看到这时gene_id还不是整数形式,所以对这列gsub()取整,命名为symbol列添加到ensemblid_log2fdc中
ensemblid_log2fdc$symbol <- gsub("\\.\\d*", "", all_deseq[,2])
4)转换id:将ENSEMBLID转为ENTREZID
entrezid <-bitr(ensemblid_log2fdc$symbol,
fromType = "ENSEMBL",
toType = c("ENTREZID"),
OrgDb = org.Hs.eg.db,
drop = TRUE)
5)因为6)中merge需要相同列名,所以将symbol列列名改为ENSEMBL
names(ensemblid_log2fdc) <- c("gene_id", "log2FoldChange", "ENSEMBL")

6)ensemblid_log2fdc 和 entrezid拼接到一起,依靠的是它们具有相同列名的列——ENSEMBL
final <- merge(ensemblid_log2fdc, entrezid)
7) 提取entrezid 和log2fdc列
library(dplyr)
entrezid_log2fdc <- dplyr::select(final, ENTREZID, log2FoldChange)
8) 用arrange()按log2fdc排序,desc()表示降序
final.sort <- arrange(entrezid_log2fdc, desc(log2FoldChange))
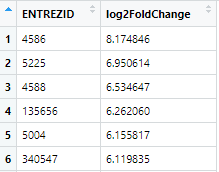
9) 构建一下要GSEA分析的数据
genelist <- final.sort$log2FoldChange
names(genelist) <- final.sort[,1]
10) 进行GSEA分析,需要entrezid,因此我前面才会进行ID转化
gsemf <- gseGO(genelist,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont="BP",
pvalueCutoff = 1)
head(gsemf)

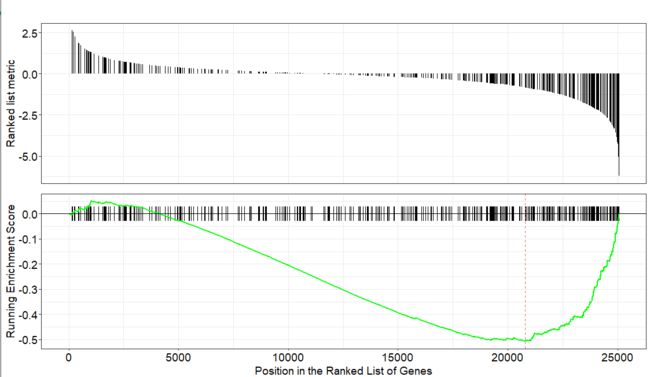
11)画出GSEA图
gseaplot(gsemf, 1)
3 可视化
1.GO富集分析结果可视化
barplot
barplot(ego, showCategory = 10) #默认展示显著富集的top10个,即p.adjust最小的10个
dotplot
dotplot(ego, showCategory = 10)
DAG有向无环图
plotGOgraph(ego) #矩形代表富集到的top10个GO terms, 颜色从黄色过滤到红色,对应p值从大到小。
igraph布局的DAG
goplot(ego)
GO terms关系网络图(通过差异基因关联)
emapplot(ego, showCategory = 30)
GO term与差异基因关系网络图
cnetplot(ego, showCategory = 5)
2.Pathway富集分析结果可视化
barplot
barplot(kk, showCategory = 10)
dotplot
dotplot(kk, showCategory = 10)
pathway关系网络图(通过差异基因关联)
emapplot(kk, showCategory = 30)
pathway与差异基因关系网络图
cnetplot(kk, showCategory = 5)
pathway映射
browseKEGG(kk, "hsa04934") #在pathway通路图上标记富集到的基因,会链接到KEGG官网