现在常用的基因定量方法包括:RPM, RPKM, FPKM, TPM。 这些表达量的主要区别是:通过不同的标准化方法为转录 本丰度提供一个数值表示,以便于后续差异分析
counts:对给定的基因组参考区域,计算比对上的read数,又称为raw count(RC)。
计数结果的差异的影响因素:落在参考区域上下限的read是否需要被统计,按照什么样的标准进行统计。
RPM方法:10^6标准化了测序深度的影响,但没有考虑转录本的长度的影响。
RPM适合于产生的read读数不受基因长度影响的测序方法,比如miRNA-seq测序,miRNA的长度一般在20-24个碱基之间。
RPKM/FPKM方法:103标准化了基因长度的影响,106标准化了测序深度的影响。
FPKM方法与RPKM类似,主要针对双末端RNA-seq实验的转录本定量。在双末端RNA-seq实验中,有左右两个对应的read来自相同的DNA片段。在进行双末端read进行比对时,来自同一DNA片段的高质量的一对或单个read可以定位到参考序列上。为避免混淆或多次计数,统计一对或单个read比对上的参考序列片段(Fragment),来计算FPKM,计算方法同RPKM。
RPKM/FPKM与RPM的区别:考虑了基因长度对read读数的影响。
RPKM与FPKM的区别:RPKM值适用于单末端RNA-seq实验数据,FPKM适用于双末端RNA-seq测序数据。
RPKM/FPKM适用于基因长度波动较大的测序方法,如lncRNA-seq测序,lncRNA的长度在200-100000碱基不等
TPM (Transcript per million):TPM的计算方法也同RPKM/FPKM类似,首先使用式2计算每个基因的表达值,去除基因长度的影响。随后计算每个基因的表达量的百分比,最后再乘以10^6,TPM可以看作是RPKM/FPKM值的百分比。
(http://www.bio-info-trainee.com/2017.html)
相当于重新标准化的文库,保证每个样本中所有TPM的总和是相同的。
TPM与RPKM/FPKM的区别:从计算公式来说,唯一的不同是计算操作的顺序,TPM是先去除了基因长度的影响,而RPKM/FPKM是先去除测序深度的影响,具体可看这篇博文,有计算步骤的详细说明;TPM实际上改进了RPKM/FPKM方法在跨样品间定量的不准确性。
TPM的使用范围与RPKM/FPKM相同。
今天在理解现在常用的基因定量方法,今天讲课的代码还没有开始,就在理解这些基础 ,不过在后续的跑代码中看到,大部分是跟之前的GSE数据分析差不多,对有一些的不理解,还需要试跑一下,而后就是再做老师布置的作业
先下载两个GSE文件,然后保存它的表达矩阵和临床信息
options(stringsAsFactors = F)#避免字符串转换成逻辑值等因子
library(GEOquery)
eSet1 <- getGEO("GSE83521",
destdir = '.',
getGPL = F)
class(eSet1)
length(eSet1)
eSet1[[1]]
class(eSet1[[1]])
exp1 <- exprs(eSet1[[1]]) #(提取表达矩阵)
exp1[1:4,1:4]
pd1 <- pData(eSet1[[1]]) #(提取临床信息)
boxplot(exp1,las=1)
eSet1[[1]]@annotation
save(pd1,exp1,file = "step1-1output.Rdata")
rm(list = ls())
eSet2 <- getGEO("GSE89143",
destdir = '.',
getGPL = F)
class(eSet2)
length(eSet2)
eSet2[[1]]
class(eSet2[[1]])
exp2 <- exprs(eSet2[[1]]) #(提取表达矩阵)
exp2[1:4,1:4]
pd2 <- pData(eSet2[[1]]) #(提取临床信息)
boxplot(exp2,las=1)
library(limma)
exp2 = log2(exp2+1)
exp2=normalizeBetweenArrays(exp2)
boxplot(exp2,las=2)
eSet2[[1]]@annotation
save(pd2,exp2,file = "step1-2output.Rdata")
接下来就是探针id注释
gpl<- getGEO('GPL19978', destdir=".")
dim(gpl)
colnames(Table(gpl)) #查一下列名
head(Table(gpl)[,c(1,2)])
ids=Table(gpl)[,c(1,2)]
ids <- ids[-c(1:2),]
ids <- ids[-c(1:13),]
save(ids,file='ids.Rdata')
在老师的发的公众号里有一个去除批次效应,就是再进行两端数据分析的时候,中间会产生误差
x1 <- exp1[rownames(exp1) %in% ids$ID,]
x2 <- exp2[rownames(exp2) %in% ids$ID,]
boxplot(x1,las=2)
boxplot(x2,las=2)
cg=intersect(rownames(x1),rownames(x2))
exp3=cbind(x1[cg,],x2[cg,])
查看boxplot
boxplot(exp3)

看到上图表达量不同,这样我们需要进行处理 进行正则表达
ibrary(limma)
## 使用 limma 的 removeBatchEffect 函数
dat[1:4,1:4]
batch <- c(rep('GSE83521',12),rep('GSE89143',6))
design=model.matrix(~group_list)
exp4 <- removeBatchEffect(dat,
batch = batch,
design = design)
dim(exp4)
{
library(limma)
fit=lmFit(exp4,design)
fit=eBayes(fit)
options(digits = 4)
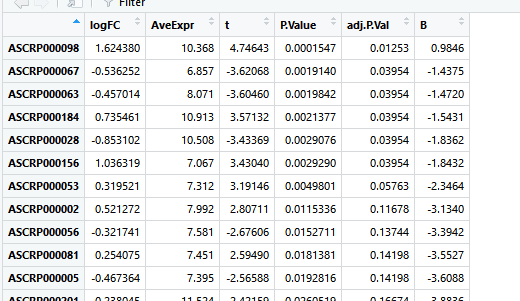
topTable(fit,coef=2,adjust='BH')
deg=topTable(fit,coef=2,adjust='BH',number = Inf)
}
library(limma)
exp4=normalizeBetweenArrays(exp4)
boxplot(exp4,las=2)

火山图
if(T){
nrDEG=deg
head(nrDEG)
attach(nrDEG)
plot(logFC,-log10(P.Value))
library(ggpubr)
df=nrDEG
df$v= -log10(P.Value) #df新增加一列'v',值为-log10(P.Value)
ggscatter(df, x = "logFC", y = "v",size=0.5)
df$g=ifelse(df$P.Value>0.05,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( df$logFC >1,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( df$logFC < -1,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
table(df$g)
df$name=rownames(df)
head(df)
ggscatter(df, x = "logFC", y = "v",size=0.5,color = 'g')
ggscatter(df, x = "logFC", y = "v", color = "g",size = 0.5,
label = "name", repel = T,
#label.select = rownames(df)[df$g != 'stable'] ,
label.select = head(rownames(deg)), #挑选一些基因在图中显示出来
palette = c("#00AFBB", "#E7B800", "#FC4E07") )
ggsave('volcano.png')
}

if(T){
up <- df[df$g=='up',]
down <- df[df$g =='down',]
x <- rbind(up,down)
x$ID <- rownames(x)
#上面是为了提取出差异基因的子集
y <- merge(x,ids,by='ID') #merge函数可以根据两列共有的内容进行合并,合并后含有共有的行名
ex_b_limma2 <- ex_b_limma[match(y$ID,rownames(ex_b_limma)),]
rownames(ex_b_limma2) <- y$circRNA
#上面的函数写的不咋好,命名变量不好命名,需要得到结果后看一下
z <- read.csv('ID.txt',sep = '\t')
tmp1 <- z[z$circRNA %in% rownames(ex_b_limma2),]
ex_b_limma3 <- ex_b_limma2[match(tmp1$circRNA,rownames(ex_b_limma2)),]
rownames(ex_b_limma3) <- tmp1$Alias
library(pheatmap)
pheatmap(ex_b_limma3,show_colnames =T,show_rownames = T)
n=t(scale(t(ex_b_limma3)))
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
pheatmap(n,show_colnames =F,show_rownames = F)
ac=data.frame(SampleType=group_list,#这步就是可以实现两个分组了
GeoDatabase=gse)
rownames(ac)=colnames(n) #将ac的行名也就分组信息 给到n的列名,即热图中位于上方的分组信息,这步很重要
pheatmap(n,show_colnames =T,
show_rownames = T,
cluster_cols = F,
annotation_col=ac,
fontsize = 8,
filename = 'deg-heatmap.png') #列名注释信息为ac即分组信息
}