一、安装软件
1、HISAT2
将reads比对到基因组上
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-Linux_x86_64.zip
unzip hisat2-2.1.0-Linux_x86_64.zip
echo 'export PATH=~/RNA-Seqruanjian/hisat2-2.1.0/bin:$PATH' >> ~/.bashrc
2、StringTie
将比对好的reads进行拼装并预计表达水平
wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.0.4.Linux_x86_64.tar.gz
tar -zvxf stringtie-2.0.4.Linux_x86_64.tar.gz
echo 'export PATH=~/RNA-Seqruanjian/stringtie-2.0.4.Linux_x86_64:$PATH' >> ~/.bashrc
3、SAM tools
课上已经用sudo apt install samtools 安装
4、gffcompare
将基因和转录本与注释进行比较,并报告有关此比较的统计数据,确定组装的转录本是否完全或部分地匹配注释的基因,并计算出多少完全是新的
wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.11.5.Linux_x86_64.tar.gz
tar -zxvf gffcompare-0.11.5.tar.gz
echo 'export PATH=~/RNA-Seqruanjian/gffcompare-0.11.5.Linux_x86_64:$PATH' >> ~/.bashrc
5、Deseq2和clusterProfiler
Deseq2:基因差异表达分析的工具,能利用RNA-Seq实验的数据,预测基因、转录本的差异表达
clusterProfiler:富集分析工具
a.安装R环境
- 首先变换软件源
sudo vi /etc/apt/sources.list
deb https://cloud.r-project.org/bin/linux/ubuntu bionic-cran35/
deb https://mirrors.tuna.tsinghua.edu.cn/CRAN/bin/linux/ubuntu/ bionic-cran35/
- 根据自己虚拟机版本,查找对应的软件源(https://cran.r-project.org/mirrors.html)
lsb_release -a #查看版本
- 加密钥
apt-get install software-properties-common dirmngr
sudo apt-key adv --keyserver keyserver.ubuntu.com --recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9
- 更新下软件源,下载R
sudo apt-get update
sudo apt-get install r-base
sudo apt-get install r-base-dev #一般上一个命令就自动安装好了这个
b.安装DEseq2和clusterProfiler
R #进入R环境
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager") #安装Bioconductor
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/") #bioconductor选择镜像
library(BiocManager) #BiocManager加载库
BiocManager::install('DEseq2')
BiocManager::install('org.Hs.eg.db') #人类注释数据库
BiocManager::install('ggplots') #画图工具包
BiocManager::install('clusterProfiler') #KEGG、GO富集分析工具包
二、找到合适的原始数据和参考基因组及其注释文件
1.在ncbi的SRA数据库找到一个合适的研究,并获取它的原始数据
- 找到了一篇关于硫酸吲哚酚通过调节经由CYP1B1产生的活性氧来刺激血管生成的课题,它这个实验研究了IS刺激和钾盐(KCl)对照,HUVEC中转录组的变化,每个做了三个样,总共6个样。
<文章链接:https://www.mdpi.com/2072-6651/11/8/454/htm;RNA-seq数据保存GSE132410>
 image.png
image.png - 下载原始数据
由于样本有点多,我们进行批量下载
a.进入SRA Run Selector ,搜索本次实验的数据SRP200940
b.勾选全部Runs的结果,点击"Accession List"键,下载得到SRR List 储存在SRR_Acc_List 文件中
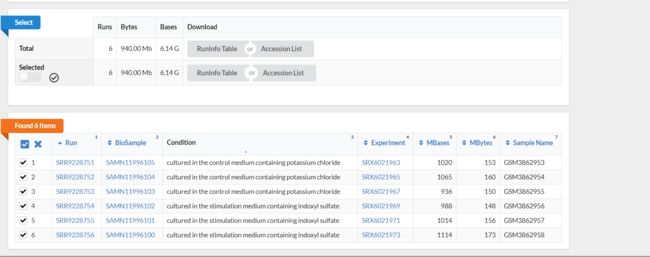
c.把SRR_Acc_List 文件上传到虚拟机中
d.批量下载
- 输入以下命令
mkdir Rna-seq-SRP200940 #存放本次实验数据
cd Rna-seq-SRP200940
prefetch --option-file SRR_Acc_List .txt
cd ~/ncbi/public/sra/
mv SRR922875* ~/Rna-seq-SRP200940/
- 得到以下结果
 image.png
image.png
2.下载参考基因组及其注释文件
- 文章中介绍他们本次实验所用的是人类参考基因组GRCh37,在genome数据库找到参考基因组文件和注释文件
 image.png
image.png - 输入以下命令
wget ftp://ftp.ensembl.org/pub/release-75/fasta/homo_sapiens/dna/Homo_sapiens.GRCh37.75.dna_sm.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz
mv Homo_sapiens.GRCh37.75.dna_sm.primary_assembly.fa.gz genome.fa.gz
mv Homo_sapiens.GRCh37.75.gtf.gz genome.gtf.gz
gunzip *.gz
mkdir GRCh37
mv genome* ./GRCh37
- 现在基本工作已经准备好
三、解压SRA文件为fastq格式
因为数据比较多,采用批量下载形式
1.新建脚本文件
vi fqdump.sh
2.输入以下脚本
#!/bin/sh
for i in *sra
do
echo $i
fastq-dump --gzip --split-files $i
done
保存退出
这里--gzip参数是为了生成压缩的gz格式fastq文件,以节省磁盘空间
3.运行脚本
sh fqdump.sh
- 结果如下
可以看出该实验是单端测序,也可以在SRA RUN select 上查看
 image.png
image.png image.png
image.png
四、用fastqc进行数据质量评价,并使用multiqc整合一下结果
fastqc *fastq.gz
multiqc .
-
可以看出该实验测序质量不错,重复reads也不多,但是还是要去掉重复序列
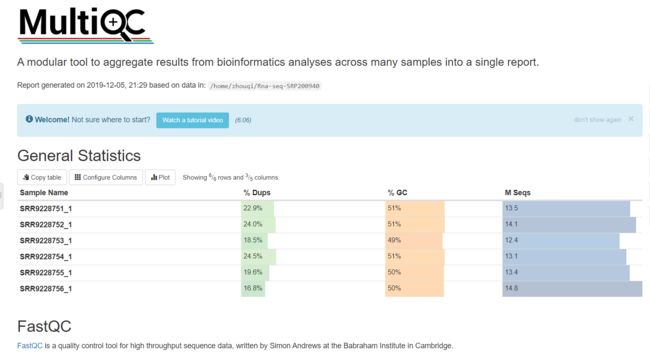 image.png
image.png
四、用Trimmomatic去重复序列
- 使用以下命令
mkdir trim_out
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228751_1.fastq.gz ./trim_out/SRR9228751_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228752_1.fastq.gz ./trim_out/SRR9228752_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228753_1.fastq.gz ./trim_out/SRR9228753_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228754_1.fastq.gz ./trim_out/SRR9228754_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228755_1.fastq.gz ./trim_out/SRR9228755_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 SRR9228756_1.fastq.gz ./trim_out/SRR9228756_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
- 批量处理
vi Trimmomatic.sh
for i in *_1.fastq.gz;
do
i=${i%_1.fastq.gz*};
echo ${i};
nohup java -jar ~/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/trimmomatic-0.38.jar SE -phred33 ${i}_1.fastq.gz ./trim_out/${i}_1.clean.fastq.gz ILLUMINACLIP:/home/zhouqi/Biosofts/Trimmomatic-0.38/Trimmomatic-0.38/adapters/TruSeq2-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75 &
done
五、用hisat2进行比对
1.给参考基因组建立索引
- 使用以下命令
hisat2-build -p 4 genome.fa genome
-p 线程数
- 也可以直接去http://ccb.jhu.edu/software/hisat2/index.shtml下载的(人和小鼠的index一般都有现成的)
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/grch37.tar.gz
2.进行比对
- 使用以下命令
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228751_1.clean.fastq.gz -S SRR9228751.sam
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228752_1.clean.fastq.gz -S SRR9228752.sam
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228753_1.clean.fastq.gz -S SRR9228753.sam
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228754_1.clean.fastq.gz -S SRR9228754.sam
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228755_1.clean.fastq.gz -S SRR9228755.sam
hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U SRR9228756_1.clean.fastq.gz -S SRR9228756.sam
- 批量比对
vi hisat2.sh
for i in *1.clean.fastq.gz;
do
i=${i%_1.clean.fastq.gz*};
echo ${i};
nohup hisat2 -p 4 --dta -x ~/Rna-seq-SRP200940/GRCh37/genome -U
${i}_1.clean.fastq.gz -S ${i}.sam &
done
-p 线程数
-x 指定基因组索引
--dta 输出转录组型报告 hisat2比对必须加上
-S 指定输出的SAM文件
-U 单端数据文件。若有多组数据,使用逗号将文件分隔。可以和-1、-2参数同时使用,Reads的长度可以不一致
-1
-2
- 成功生成sam格式文件
 image.png
image.png
六、利用samtools对sam格式的比对文件进行处理
1.为参考基因组建立索引
- 输入下列命令
samtools faidx genome.fna
2.将sam格式转换为二进制格式bam
- 输入下列命令
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228751.bam SRR9228751.sam
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228752.bam SRR9228752.sam
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228753.bam SRR9228753.sam
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228754.bam SRR9228754.sam
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228755.bam SRR9228755.sam
samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o SRR9228756.bam SRR9228756.sam
- 批量处理
vi view.sh
for i in *.sam;
do
i=${i%.sam*};
echo ${i};
nohup samtools view -bhS -t ~/Rna-seq-SRP200940/GRCh37/genome.fa.fai -@ 2 -o ${i}.bam ${i}.sam &
done
-b output BAM 默认下输出的是SAM格式,这参数设置输出为BAM格式
-h print header for the SAM output 默认下输出的 sam 格式文件不带 header,该参数设定输出sam文件时带 header 信息
-S input is SAM 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。
-t file list of reference names and lengths (force -S) 使用一个list文件来作为header的输入
-@ Number of additional threads to use [0] 指使用的线程数
-o FILE output file name [stdout] 输出文件的名称
-
得到下列结果

3.对 bam 文件中的内容进行排序
- 输入下列命令
samtools sort -@ 4 -o SRR9228751.sort.bam SRR9228751.bam
samtools sort -@ 4 -o SRR9228752.sort.bam SRR9228752.bam
samtools sort -@ 4 -o SRR9228753.sort.bam SRR9228753.bam
samtools sort -@ 4 -o SRR9228754.sort.bam SRR9228754.bam
samtools sort -@ 4 -o SRR9228755.sort.bam SRR9228755.bam
samtools sort -@ 4 -o SRR9228756.sort.bam SRR9228756.bam
- 批量处理
vi sort.sh
for i in *.bam;
do
i=${i%.bam*};
echo ${i};
nohup samtools sort -@ 4 -o ${i}.sort.bam ${i}.bam &
done
- 得到下列结果
 image.png
image.png
4.可视化比对结果
a、先对排序后的bam文件进行索引
samtools index SRR9228751.sort.bam
samtools index SRR9228752.sort.bam
samtools index SRR9228753.sort.bam
samtools index SRR9228754.sort.bam
samtools index SRR9228755.sort.bam
samtools index SRR9228756.sort.bam
b、可视化结果
samtools tview SRR9228751.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
samtools tview SRR9228752.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
samtools tview SRR9228753.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
samtools tview SRR9228754.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
samtools tview SRR9228755.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
samtools tview SRR9228756.sort.bam ~/Rna-seq-SRP200940/GRCh37/genome.fa
七、利用StringTie进行转录本组装,和量化基因表达
1.对样本进行组装
比对上的reads将会被呈递给StringTie进行转录本组装,StringTie单独的对每个样本进行组装,在组装的过程中顺带估算每个基因及isoform的表达水平
- 输入下列命令
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228751.gtf -l SRR9228751 SRR9228751.sort.bam
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228752.gtf -l SRR9228752 SRR9228752.sort.bam
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228753.gtf -l SRR9228753 SRR9228753.sort.bam
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228754.gtf -l SRR9228754 SRR9228754.sort.bam
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228755.gtf -l SRR9228755 SRR9228755.sort.bam
stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o SRR9228756.gtf -l SRR9228756 SRR9228756.sort.bam
-p 线程(CPU)数 (default: 1)
-G 参考序列的基因注释文件 (GTF/GFF3)
-l 输出转录本的名称前缀(默认为MSTRG)
- 或者使用脚本批量组装
vi stringtie1.sh
for i in *sort.bam;
do
i=${i%.sort.bam*};
echo ${i};
nohup stringtie -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf
-o ${i}.gtf -l ${i} ${i}.sort.bam &
done
- 结果如下
 image.png
image.png
2.将所有转录本合并
所有的转录本都被呈递给StringTie的merge函数进行merge,这一步是必须的,因为有些样本的转录本可能仅仅被部分reads覆盖,无法被StringTie组装出来。merge步骤可以创建出所有样本里面都有的转录本
<链接:https://www.jianshu.com/p/1f5d13cc47f8>
- 输入下列命令
创建mergelist
vi mergelist.txt #需要包含之前output.gtf文件的路径
SRR9228751.gtf
SRR9228752.gtf
SRR9228753.gtf
SRR9228754.gtf
SRR9228755.gtf
SRR9228756.gtf
- 转录本合并
stringtie --merge -p 4 -G ~/Rna-seq-SRP200940/GRCh37/genome.gtf -o stringtie_merged.gtf mergelist.txt
-p 线程(CPU)数 (default: 1)
-G
3.检测相对于注释基因组,转录本的组装情况
使用gffCompare实用程序来确定有多少组合的转录本完全或部分匹配带注释的基因,并计算出有多少是完全新颖的
- 输入下列命令
gffcompare -r ~/Rna-seq-SRP200940/GRCh37/genome.gtf -G -o merged stringtie_merged.gtf
-r 参考转录本的注释信息
-G 比对所有转录本
-0 指定要用于gffcompare将创建的输出文件的前缀
- 结果如下

 image.png
image.pnggffcmp.annotated.gtf 这里面向每个转录本添加一个"类代码"和来自参考注释文件的转录本的名称,这使用户能够快速检查预测的转录本与参考基因组的关系。
精度:显示与参考基因组重叠的gene的比例
gffcmp.stats 包含不同基因特征的灵敏度和精度
灵敏度:参考基因组中正确重建的的基因比例
 image.png
image.png
4.重新组装转录本并估算基因表达丰度
- 输入下列命令
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228751/SRR9228751.gtf SRR9228751.sort.bam
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228752/SRR9228752.gtf SRR9228752.sort.bam
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228753/SRR9228753.gtf SRR9228753.sort.bam
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228754/SRR9228754.gtf SRR9228754.sort.bam
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228755/SRR9228755.gtf SRR9228755.sort.bam
stringtie –e –B -p 4 -G stringtie_merged.gtf -o ballgown/SRR9228756/SRR9228756.gtf SRR9228756.sort.bam
- 批量处理
vi chongzuzhuang.sh
for i in *.sort.bam;
do
i=${i%.sort.bam*};
echo ${i};
nohup stringtie -e -B -p 8 -G stringtie_merged.gtf -o ballgown/${i}/${i}.gtf ${i}.sort.bam &
done
-e 只对参考转录本进行丰度评估 (requires -G)
-G 参考序列的基因注释文件 (GTF/GFF3)
-B 在输出的GFT同目录下输出Ballgown table 文件
八、利用DEseq2进行基因差异表达分析
1.stringtie输出的结果为ballgown所需要的格式,需要转换为deseq2需要的表格
a.下载一个python脚本prepDE.py
wget http://ccb.jhu.edu/software/stringtie/dl/prepDE.py
b.转换格式
python2 ./prepDE.py - ballgown
- 生成两个csv文件
gene_count_matrix.csv
transcript_count_matrix.csv
2.用DESeq2分析
R
library(DESeq2)
#导入数据
CountMatrix1<-read.csv("gene_count_matrix.csv",sep=",",row.names="gene_id")
##修改列名
names(CountMatrix1)<-c("ctrlrep1","ctrlrep2","ctrlrep"," ISrep1","ISrep2","ISrep3")
#设置样本信息矩阵,包括处理信息:实验组ctrlrep vs. 对照组ISrep,每个有3个
ColumnData<- data.frame(row.names=colnames(CountMatrix1),samName=colnames(CountMatrix1), condition=rep(c("ctrlrep","ISrep"),each=3))
#生成DESeqDataSet数据集
dds<-DESeqDataSetFromMatrix(countData = CountMatrix1, colData = ColumnData, design = ~ condition)
#DESeq差异表达计算
dds<-DESeq(dds)
#生成差异表达结果
res<-results(dds)
summary(res)
#查看总结信息(表达上调,下调等)
head(res)
#统计padj(adjusted p-value)小于0.05的数目
table(res$padj <0.05)
#统计padj(adjusted p-value)小于0.05的数目
res<- res[order(res$padj),]#按padj排序
resdata <- merge(as.data.frame(res), as.data.frame(counts(dds, normalized=TRUE)),by="row.names",sort=FALSE)
write.csv(resdata,file = "SRP200940.csv")
#输出结果到csv文件
deg <- subset(res, padj <= 0.01 & abs(log2FoldChange) >= 1) #筛选显著差异表达基因(padj小于0.01且FoldChange绝对值大于2)
summary(deg) #查看筛选后的总结信息
write.csv(deg, "SRP200940.deg.csv") #将差异表达显著的结果输出到csv文件
3.用ggplots作图
library(ggplot2)
volcano<- ggplot(resdata, aes(x= log2FoldChange, y= -1*log10(padj)))
#x轴为log2FC;y轴为-log(padj)
threshold<-as.factor(resdata$padj <= 0.01 & abs(resdata$log2FoldChange) >= 2)
#筛选条件(阈值):绝对log2FC大于2,并且padj<0.01
p1<-volcano+geom_point(aes(color=threshold))
p1
#加上各数据点信息
p2<-p1+scale_color_manual(values=c("grey","red"))
p2
#更改散点颜色
p3<-p2+geom_hline(yintercept=2,linetype=3)+geom_vline(xintercept=c(-2,2),linetype=3)
p3
#加上水平和垂直线,标识阈值选择范围
p4<-p3+theme(axis.line=element_line(colour="black"),panel.background = element_rect(fill = "white"))
p4
#修改图片背景填充颜色,坐标轴线条颜色
degs <- subset(resdata, padj <= 0.01 & abs(log2FoldChange)>= 2)
p5<-p4+geom_text(aes(label=degs$Row.names),hjust=1, vjust=0,data = degs)
p5
#绘制P-value图
hist(deg$pvalue,breaks=10,col="grey",xlab="p-value")
#绘制MA图
plot(deg$log2FoldChange,-log2(deg$padj),col=ifelse(abs(deg$log2FoldChange) >= 2 & abs(deg$padj) <= 0.05,"red","black"),xlab="log2FoldChange",ylab="-log2Pvalue")
九、利用clusterProfiler进行基因差异表达分析
把SRP200940.deg.csv下载用excel筛选只剩下id和foldchange
并在https://david.ncifcrf.gov/conversion.jsp进行id转换为genesymbol
library(org.Hs.eg.db) #人类注释数据库
library(ggplot2) #画图工具包
library(clusterProfiler) #KEGG、GO富集分析工具包
#读入差异表达基因列表,并且需要标题行
geneList <- read.table("SRP200940.deg.txt",header=TRUE)
#将基因列表的gene Symbol 转换成 entrez ID
geneID <- bitr(as.character(geneList$geneSymb),fromType="SYMBOL",toType=c("ENTREZID"),OrgDb=org.Hs.eg.db)
#差异表达基因的功能富集分析
KEGGenrich <- enrichKEGG(geneID$ENTREZID,organism='hsa',pvalueCutoff = 0.05)
write.csv(summary(KEGGenrich),"GeneEnrichment_results.csv")
#kegg柱状图绘制
pdf("Gene_KEGGenrichment_barplot.pdf")
barplot(KEGGenrich,title="KEGG enrichment")
dev.off()
#GO 富集分析
enrichBP <- enrichGO(geneID$ENTREZID,OrgDb=org.Hs.eg.db,ont="BP",pvalueCutoff = 0.05) #分析生物学过程方面
enrichMF <- enrichGO(geneID$ENTREZID,OrgDb=org.Hs.eg.db,ont="MF",pvalueCutoff = 0.05) #分析分子功能方面
enrichCC <- enrichGO(geneID$ENTREZID,OrgDb=org.Hs.eg.db,ont="CC",pvalueCutoff = 0.05) #分析细胞组成方面
#查看富集结果的个数:
dim(enrichBP)
dim(enrichMF)
dim(enrichCC)
#柱状图绘制
#BP
pdf("Gene_GOenrichBP_barplot.pdf")
barplot(enrichBP,title="GOenrichBP")
dev.off()
#MF
pdf("Gene_GOenrichMF_barplot.pdf")
barplot(enrichMF,title="GOenrichMF")
dev.off()
#CC
pdf("Gene_GOenrichCC_barplot.pdf")
barplot(enrichCC,title="GOenrichCC")
dev.off()
·: