modeller建模(单模板建模、多模板建模)
- 题目要求
1、采用modeller对给定序列进行建模,单模板和多模板方法各建3个模型,每种方法选择一个最优的模型,最后进行比较,选出最优的模型(怎样比较两个模型的好坏,可以从哪些角度去看)。并说明理由为何选该模型。
2、单模板结果与Swiss-model的结果进行简单的比较,说明异同。(其实这里还是涉及了模型之间比较的问题)
3、构建的模型都要在molprobity和SAVES中的工具进行评估,并对评估结果进行注释(说明了什么?)
4、请对实验流程、所用脚本、结果以及重要的参数选择进行记录和说明。
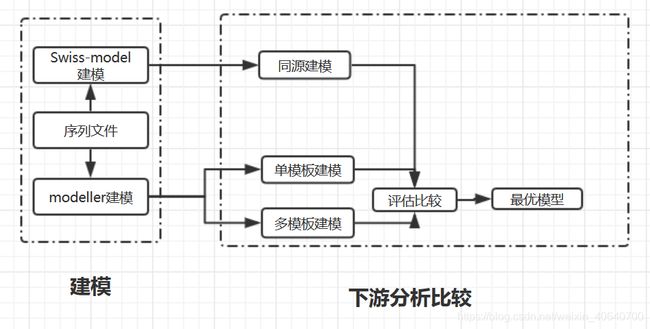
图:实验流程 1
- 操作过程记录及结果
(一)前期准备
1、modeller软件的下载
https://salilab.org/modeller/download_installation.html

本次实验选择版本为Windows(64-bit)
安装注册码:MODELIRANJE
2、序列文件
>jgi|Cormi1|1163|CCM_01166m.01
MMGLGSVTFDEHNPNSEKGRENDARTSKSGLDYSPLPRLTGKSLSMGILVSMGGLIFGYDTGQISGFLEMPDFLARFGQLDSNGNAYFSRVRSGLIVSLLSIGTLIGALVAAPIADRFGRRPSISFWALMTAVGFVIQISADTAWTQIMMGRFVAGLGVGALSLLVPMYQAETAPAWIRGAMVCAYQLFITMGIFLAACFNFGTYHHQMHNSSSWRIVIGLGWIWTVVLGVGILFFPETPRFDYRRGKTEEAIKTLCDVHGAPRNHYIVHTQVAEIESKLHAESKIKKGHIAEFTSMLKAPRMLYRLSLGVALQSLQQLTGANFFFYYGVSIFKSVNIDSFITQIILNTINFLVTFIGLYIVEHFGRRKSLIAGSVWMFICFLIFASVGHFSLDYNNPENTPTPGIVLIVFAALFILGFATTWGPMIWTIQAELFPSRYRAKGMAISTAANWIWNFCIGFFTPFITGAIDYRYGYVFAACNFVAGFVIYFFVIEGQGRTIEEIDTMYLERVTPWKSAEWVAPDADQMEQIRRKAGIDVEALAAQNDEHSKIAGDPQGGVRVDGTNMDHVE
(二)Swiss-model同源建模
登录SWISS-MODEL网页,https://www.swissmodel.expasy.org/,提交序列,等待模型的预测结果。得到建模结果:https://www.swissmodel.expasy.org/interactive/2wz5cB/models/,取序列相似性为31.81%的模型(相似性越高,同源建模的准确率越高)。该模型以6h7d.1.A序列为模板。可以观察到模板序列与提交的目标蛋白序列并没有完全覆盖,首尾部分大概都有45bp长度缺失。
(三)运用modeller建模
单模板建模和多模板建模的异同:
单模板建模是通过序列比对的方法,找到与目标序列相似度最高的一条序列作为模板(序列相似=>结构相似),再在此基础上进行优化。
多模板建模是通过多序列比对,这些序列中的保守区域的结构特征很容易确定,对于差异的片段,在多条序列片段中找到与目标序列片段最主导(Dominant)的结构。然后将这些碎片的结构拼接成一个完整的目标序列结构。
【操作参考说明文档】
单模板建模:https://salilab.org/modeller/tutorial/basic.html
多模板建模:https://salilab.org/modeller/tutorial/advanced.html
3.1、单模板建模
3.1.2、选择和目标蛋白序列相关的结构
1、将序列文件修改为.ali格式文件。命名为seq.ali。
>P1;CCM_01166m
sequence:CCM_01166m:::::::0.00: 0.00
MMGLGSVTFDEHNPNSEKGRENDARTSKSGLDYSPLPRLTGKSLSMGILVSMGGLIFGYDTGQISGFLEMPDFLARFGQLDSNGNAYFSRVRSGLIVSLLSIGTLIGALVAAPIADRFGRRPSISFWALMTAVGFVIQISADTAWTQIMMGRFVAGLGVGALSLLVPMYQAETAPAWIRGAMVCAYQLFITMGIFLAACFNFGTYHHQMHNSSSWRIVIGLGWIWTVVLGVGILFFPETPRFDYRRGKTEEAIKTLCDVHGAPRNHYIVHTQVAEIESKLHAESKIKKGHIAEFTSMLKAPRMLYRLSLGVALQSLQQLTGANFFFYYGVSIFKSVNIDSFITQIILNTINFLVTFIGLYIVEHFGRRKSLIAGSVWMFICFLIFASVGHFSLDYNNPENTPTPGIVLIVFAALFILGFATTWGPMIWTIQAELFPSRYRAKGMAISTAANWIWNFCIGFFTPFITGAIDYRYGYVFAACNFVAGFVIYFFVIEGQGRTIEEIDTMYLERVTPWKSAEWVAPDADQMEQIRRKAGIDVEALAAQNDEHSKIAGDPQGGVRVDGTNMDHVE*
2、运行build_profile.py文件。
修改build_profile.py文件中的参数。
| (略去开头) #-- Read in the target sequence/alignment aln = alignment(env) aln.append(file='seq.ali', alignment_format='PIR', align_codes='ALL')
#-- Convert the input sequence/alignment into # profile format prf = aln.to_profile()
#-- Scan sequence database to pick up homologous sequences prf.build(sdb, matrix_offset=-450, rr_file='${LIB}/blosum62.sim.mat', gap_penalties_1d=(-500, -50), n_prof_iterations=5, check_profile=False, max_aln_evalue=1, gaps_in_target=False) #略去结尾 |
修改aln.append()中的file参数为我们的目标序列文件名seq.ali。
修改参数max_aln_evalue为1(这样在输出文件buildprofile.prf中会有更多的检索结果)。
将seq.ali文件和build_profile.py等文件放置在同一路径下。在该路径下打开cmd。

运行指令:
Python build_profile.py > build_profile.log
得到结果文件:

其中buildprofile.prf文件中存放有检索模板,可以打开查看。
|
|

其中第一行是我们的目标序列,下面的就是检索出来的模板。
将上述模板检索结果保存为.txt文件,用excel处理,根据第12列的数值大小排序,选择evalue较小的4条序列(evalue=0)。
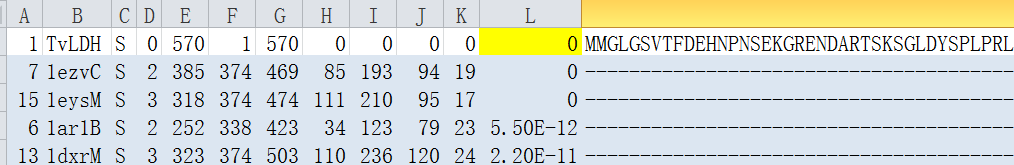
记录下pdb号,为“1ezvC”、“1eysM”、“1ar1B”、“1dxrM”。
从pdb数据库(http://www.rcsb.org/)中下载以上模板的pdb.文件,备用。

3、比较结构与序列之间的相似性。(compare.py)
将模板的pdb文件和compare.py放置在同一文件夹下。
修改compare.py文件。
| from modeller import * env = environ() aln = alignment(env) for (pdb, chain) in (('1ezv', 'C'), ('1eys', 'M'),('1dxr', 'M') ): m = model(env, file=pdb, model_segment=('FIRST:'+chain, 'LAST:'+chain)) aln.append_model(m, atom_files=pdb, align_codes=pdb+chain) aln.malign() aln.malign3d() aln.compare_structures() aln.id_table(matrix_file='family.mat') env.dendrogram(matrix_file='family.mat', cluster_cut=-1.0) |
由于“1ar1B”添加进去程序报错,未找到错误原因,遂删去“1ar1B”,接着程序可以正常运行。
在文件夹路径下打开cmd,输入指令。
Python compare.py >compare.log
打开结果文件compare.log
| 1ezvC @21eysM @21dxrM @2 1ezvC @2 385 4 4 1eysM @2 1 318 187 1dxrM @2 1 59 323 Weighted pair-group average clustering based on a distance matrix:
.---------------------------------------------------------- 1ezvC @2.3 99.0000 | | .--- 1eysM @2.2 41.0000 | | .------------------------------------------------------------ 1dxrM @2.0
+----+----+----+----+----+----+----+----+----+----+----+----+ 101.3200 90.8800 80.4400 70.0000 59.5600 49.1200 38.6800 96.1000 85.6600 75.2200 64.7800 54.3400 43.9000 |
暂时选择1ezvC作为模板(至于如何选择,官网上的筛选条件我没怎么看懂)。
4、将目标序列同模板比较。(align2d.py)
| from modeller import * env = environ() aln = alignment(env) mdl = model(env, file='1ezv', model_segment=('FIRST:C','LAST:C')) aln.append_model(mdl, align_codes='1ezvC', atom_files='1ezv.pdb') aln.append(file='seq.ali', align_codes='CCM_01166m') #CCM_01166m是seq.ali文件第二行的序列名 aln.align2d() aln.write(file='align.ali', alignment_format='PIR') aln.write(file='align.pap', alignment_format='PAP') |
修改文件中标记处。
将模板的lezv.pdb文件同align2d.py和seq.ali(目标序列文件)文件放在同一个文件夹下。

在align2d.py文件所在路径下打开cmd。运行指令。
Python align2d.py >align2d.log
生成结果文件:

5、单模板建模(model-single.py)
打开model-single.py文件。
| from modeller import * from modeller.automodel import * #from modeller import soap_protein_od env = environ() a = automodel(env, alnfile='align.ali', knowns='1ezvC', sequence='CCM_01166m', assess_methods=(assess.DOPE, #soap_protein_od.Scorer(), assess.GA341)) a.starting_model = 1 a.ending_model = 6 #最终生成的模型的数目,可修改 a.make() |
修改文件标记处。
上一步产生的align.ali和model-single.py放在同一个文件夹下。
![]()
在model-single.py文件所在路径下打开cmd。运行指令。
Python model-single.py >model-single.log
生成模型文件:

模型的详细信息,参见model-single.log文件
| >> Summary of successfully produced models: Filename molpdf DOPE score GA341 score ---------------------------------------------------------------------- CCM_01166m.B99990001.pdb 4224.62891 -55072.33594 0.04189 CCM_01166m.B99990002.pdb 3910.86230 -53359.83984 0.04221 CCM_01166m.B99990003.pdb 3997.98511 -54259.32031 0.06955 CCM_01166m.B99990004.pdb 3903.93872 -53906.61328 0.07631 CCM_01166m.B99990005.pdb 3726.95410 -54619.51953 0.07964 CCM_01166m.B99990006.pdb 3893.26294 -53613.23828 0.03485 |
选择其中最优的模型CCM_01166m.B99990005.pdb(DOPE score最低且GA341 score最高)进行下游分析。
3.2、多模板建模
1、多模板的选择
(1)方案一:序列比对,找多条相似序列(序列相似=>结构相似)。
使用NCBI的blast比对工具。选择Protein Data Bank proteins Database。
检索结果:
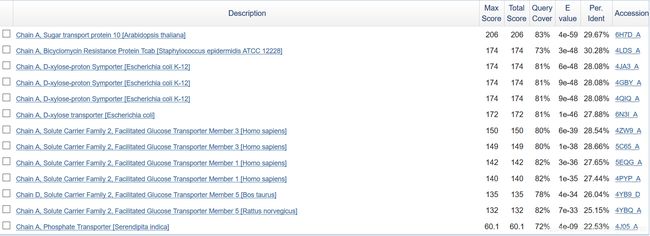 如下图。
如下图。
选择6H7D_A、4LDS_A、4JA3_A、4GBY_A、4QIQ_A模板建模(他们都含有Sugar_tr结构域)。

(2)方案二:找单模板建模时匹配到的最优模板1ezvC同一家族的蛋白质模板。
到pfam数据库中,在数据库的搜索框中输入1ezv的id号。
http://pfam.xfam.org/structure/1ezv
1ezvC所属的家族为Cytochrome_B和Cytochrome_B_C。

选择1EZV_C、1KB9_C、1KYO_C、1KYO_N、1P84_C、2IBZ_C、3CX5_C、3CX5_N、3CXH_C、3CXH_N、4PD4_C作为模板建模。
综合方案一和方案二的模板,这些序列都用来进行多模板建模。
到pdb数据库(http://www.rcsb.org/)中下载模板的pdb文件,为下一步的操作做准备。
2、模板序列的多序列比对(salign.py)
| from modeller import *
log.verbose() env = environ() env.io.atom_files_directory = './'
aln = alignment(env) for (code, chain) in (('1ezv', 'C'), ('1kb9', 'C'), ('1kyo', 'C'),('1kyo', 'N'),('1p84', 'C'),('2ibz', 'C'),('3cx5', 'C'),('3cx5', 'N'),('3cxh', 'N'),('3cxh', 'C'),('4pd4', 'C'), ('6h7d', 'A'),('4gby', 'A'),('4gbz', 'A')): mdl = model(env, file=code, model_segment=('FIRST:'+chain, 'LAST:'+chain)) aln.append_model(mdl, atom_files=code, align_codes=code+chain)
for (weights, write_fit, whole) in (((1., 0., 0., 0., 1., 0.), False, True), ((1., 0.5, 1., 1., 1., 0.), False, True), ((1., 1., 1., 1., 1., 0.), True, False)): aln.salign(rms_cutoff=3.5, normalize_pp_scores=False, rr_file='$(LIB)/as1.sim.mat', overhang=30, gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0, dendrogram_file='temple.tree', alignment_type='tree', # If 'progresive', the tree is not # computed and all structues will be # aligned sequentially to the first feature_weights=weights, # For a multiple sequence alignment only # the first feature needs to be non-zero improve_alignment=True, fit=True, write_fit=write_fit, write_whole_pdb=whole, output='ALIGNMENT QUALITY')
aln.write(file='temple.pap', alignment_format='PAP') aln.write(file='temple.ali', alignment_format='PIR')
aln.salign(rms_cutoff=1.0, normalize_pp_scores=False, rr_file='$(LIB)/as1.sim.mat', overhang=30, gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0, dendrogram_file='1is3A.tree', alignment_type='progressive', feature_weights=[0]*6, improve_alignment=False, fit=False, write_fit=True, write_whole_pdb=False, output='QUALITY') |
根据实际情况,修改salign.py文件的划线处。
3、目标序列同模板结构比对(align2d_mult.py)
| from modeller import *
log.verbose() env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib')
# Read aligned structure(s): aln = alignment(env) aln.append(file='temple.ali', align_codes='all') #temple.ali是上一步操作的结果文件 aln_block = len(aln)
# Read aligned sequence(s): aln.append(file='seq.ali', align_codes='CCM_01166m') #seq.ali是目标序列文件
# Structure sensitive variable gap penalty sequence-sequence alignment: aln.salign(output='', max_gap_length=20, gap_function=True, # to use structure-dependent gap penalty alignment_type='PAIRWISE', align_block=aln_block, feature_weights=(1., 0., 0., 0., 0., 0.), overhang=0, gap_penalties_1d=(-450, 0), gap_penalties_2d=(0.35, 1.2, 0.9, 1.2, 0.6, 8.6, 1.2, 0., 0.), similarity_flag=True)
aln.write(file='seq-mult.ali', alignment_format='PIR') aln.write(file='seq-mult.pap', alignment_format='PAP') |
4、建立模型(model_mult.py)
| from modeller import * from modeller.automodel import *
env = environ() a = automodel(env, alnfile='seq-mult.ali', knowns=('1ezvC','1kb9C','1kyoN','1p84C','2ibzC','3cx5N','3cxhC','4pd4C','6h7dA','4gbyA','4gbzA'), sequence='CCM_01166m') a.starting_model = 1 a.ending_model = 2 a.make() |
建造了的两个模型。
![]()
5、模型评估(evaluate_model.py)
| from modeller import * from modeller.scripts import complete_pdb
log.verbose() # request verbose output env = environ() env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file mdl = complete_pdb(env, 'CCM_01166m.B99990002.pdb')
# Assess with DOPE: s = selection(mdl) # all atom selection s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='CCM_01166m2.profile', normalize_profile=True, smoothing_window=15) |
根据实际情况修改上面划线处的参数。
评估结果:
DOPE score
CCM_01166m.B99990001.pdb:-60330.765625
CCM_01166m.B99990002.pdb:-60692.351562
第二个模板的DOPE打分值更小一点,所以多模板建模结果选择第二个模板作为最优模板。
三、结果讨论和分析
(一)模型优化
主要对多模板建模的模型尝试进行优化。

可以看出同单模板建模的模型相比,多模板在0~100区间内明显优于单模板。在410~435区间内的值略高。
于是尝试在这个区间范围内对多模板建模模型进行环区优化。
loop_refine.py
| # Loop refinement of an existing model from modeller import * from modeller.automodel import *
log.verbose() env = environ()
# directories for input atom files env.io.atom_files_directory = './'
# Create a new class based on 'loopmodel' so that we can redefine # select_loop_atoms (necessary) class MyLoop(loopmodel): # This routine picks the residues to be refined by loop modeling def select_loop_atoms(self): # 10 residue insertion return selection(self.residue_range('410', '435'))
m = MyLoop(env, inimodel='CCM_01166m.B99990002.pdb', # initial model of the target sequence='CCM_01166m') # code of the target
m.loop.starting_model= 1 # index of the first loop model m.loop.ending_model = 10 # index of the last loop model m.loop.md_level = refine.very_fast # loop refinement method; this yields # models quickly but of low quality; # use refine.slow for better models
m.make() |
(根据实际情况,修改上方文件划线处)
一共建立了10个模型,其中第二个优化模型的DOPE 值最小(-52333.929688),选择该模型进行模型评估,绘图。

可以看出环区优化之后,橙色区域的DOPE打分值有明显下降。
但是,后来用VMD可视化查看模型结构时,发现在该模型在1~85区间内有很长的尾巴,无结构。(这也可以解释上图中,为什么多模板模型同单模板模型比较,1~100区间内DOPE 的打分值惊人的小)

于是决定重新进行多模板建模。
将序列1~85位氨基酸序列取出,使用的NCBI的blastp搜索同源结构。
找到同源模板6h7d_A,4gby_A,4gbz_A,下载pdb文件,将模板添加到多模板建模的文件中,重新建模评估。
得到下方的结果。

在vmd中查看,优化后的模型没有很长的无序结构,基本满足要求。

(二)模型可视化比较
将Swiss-model、modeller单模板建模、modeller多模板建模的模型在vmd的RMAD的工具中进行比较。可以计算出模型的偏差,如下图所示。
其中46~496为序列的MFS结构域区段。
参见uniprot中对于该实验目标序列CCM_01166的介绍:
https://www.uniprot.org/uniprot/G3J3I1
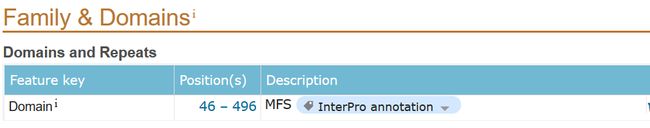

可以看出,与Swiss model建模结果比较,modeller 单模板建模和双模板建模的模板与之偏差很大(>20)。
【单模板建模比较】
Swiss model建模和modeller单模板建模,都采用了单模板建模的方法。但是在具体的操作过程还是存在很多差异。
首先Swiss model和modeller搜索到的最优模板不同。对于本次实验,Swiss model的最优模板是6h7dA;但是modeller的最优模板是1ezvC。
其次,Swiss model和modeller处理未匹配的序列的方式不一样,Swiss model将模板与目标序列不匹配的区段切除。modeller将不匹配区段以loop方式保留。
最后,在使用方式上,个人认为modeller更加灵活,可扩展性更强,我可以在建模pipline的任何一个环节进行个性化的调整。
(三)模型评估
1、Swiss-model建模评估结果
(1)moplrobity评估结果
(2)SAVES平台评估
- 拉氏图
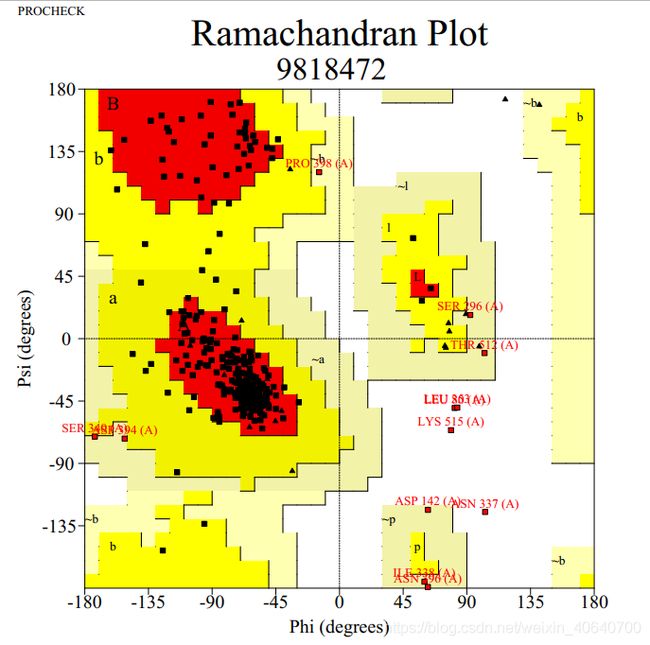
其中仅有90.8%(377/415)的氨基酸落在the most favoured regions,按照一般的标准(一个比较好的模型应该有90%以上的氨基酸落在 the most favoured regions),该模型符合要求,但是由于目标序列的长度是570个氨基酸,该模型在构造的过程中切掉了一部分,所以在后续使用时需要额外注意。
- Verify 3D (检验结果3D结果的兼容性)
仅有64.36% 残基的平均3D-1D 值>= 0.2(一般标准是至少80%的氨基酸残基的平均3D-1D 值>= 0.2),所以该模型的三维结构不兼容,需要进一步优化。

- ERRAT(统计非键相互作用)
Quality Factor: A: 93.3619
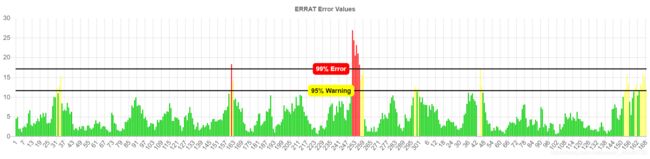
在253~259区间中结构出错。可以考虑模型局部优化。
- WHATCHECK :立体化学
该结构存在侧链平面角度问题;连接到平面外的芳香环问题;异常短的原子间距离问题;骨架构造Z值非常低问题;HIS, ASN, GLN 侧链翻转问题;平均B因子错误;
2、modeller单模板建模评估结果
(1)moplrobity评估

(2)SAVES平台评估
- 拉氏图

其中仅有81.5%(402/570)的氨基酸落在the most favoured regions,按照一般的标准(一个比较好的模型应该有90%以上的氨基酸落在 the most favoured regions),该模型不符合要求,需要优化。
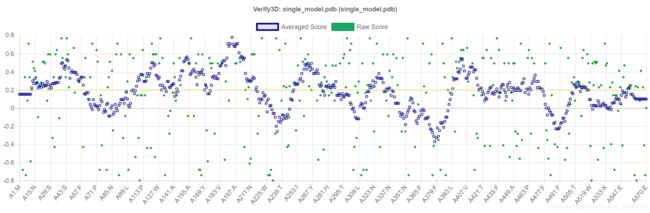
- Verify 3D (检验结果3D结果的兼容性)
仅有49.12% 残基的平均3D-1D 值>= 0.2(一般标准是至少80%的氨基酸残基的平均3D-1D 值>= 0.2),所以该模型的三维结构不兼容,需要进一步优化。
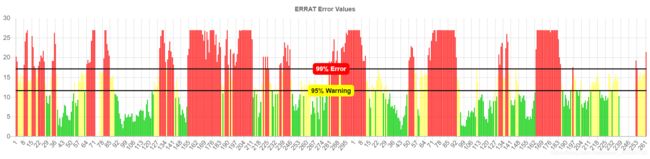
- ERRAT(统计非键相互作用)
Quality Factor: A: 37.5
- WHATCHECK :立体化学
该结构模型存在异常短的原子间距离问题;骨架构造z值非常低的问题;平均B因子错误;HIS, ASN, GLN侧链翻转问题;
3、modeller多模板建模评估结果
(1)moplrobity评估

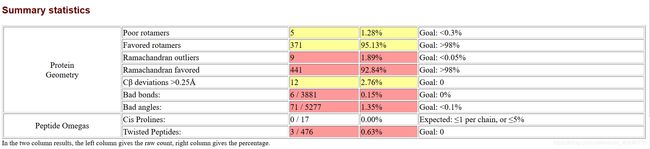
ramachandran outliers和ramachandran favored 可以和PROCHECK评估结果的ramachandran plot 参照着看。ramachandran outliers就相当于落在白色区域内的氨基酸的数量,ramachandran favored相当于落在红色区域内的氨基酸数量。一个好的结构模型,我们希望有更多的氨基酸落在红色区域内(>98%)。但是从我们结构的实际结果看,ramachandran favored的值和期望值有很大差异。该模型不是最优模型,需要进一步优化。
rotamer的中文意思是旋转异构体,即由于绕单键旋转受阻碍而不能轻易互相转换的一组构象异构体。对于同一个分子而言,可以有多个构象变化。每个构象都有自己的势能。随着转动角度的变化,势能也起伏变化,呈现出几个极小值。具有极小值势能的几个构象互为构象异构体。
poor rotamers指的是构象异构体中势能最大的结构。
favor rotamers指的是构象异构体中势能最小的结构。
一般而言,分子中favor rotamer的比例大于98%,poor rotamer的比例小于0.3%,这样的模型是稳定的合理的结构。但是就本模型的实际情况而言,poor rotamer的比例为6.01%,远远大于0.3%,favor rotamer的比例小于标准值。因此该模型需要进一步的修正。
脯氨酸是环状的亚氨酸,容易形成顺式肽链。常规情况下,肽键大多是反式结构,形成稳定的肽平面。一个结构中顺式肽键含量多,会影响结构的稳定性。因此将顺式脯氨酸的含量也作为模型好坏的评价指标之一。但是,从上表实际 的结果可以看出,该模型不存在这类问题。
总体来说,这个模型的结构评估结果非常的糟糕
(2)SAVES平台评估
- 拉氏图(检验蛋白质的构象是否合理)

其中仅有81.5%(402/570)的氨基酸落在the most favoured regions,按照一般的标准(一个比较好的模型应该有90%以上的氨基酸落在 the most favoured regions),该模型不符合要求,需要优化。
- Verify 3D
verify 3D是通过根据原子模型的位置和环境(alpha、beta、loop、polar、nonpolar等)分配结构类,并将结果与良好结构进行比较,确定原子模型(3d)与其自身氨基酸序列(1d)的兼容性的工具。
从下图的分析结果可以看出:仅有60.88% 残基的平均3D-1D 值>= 0.2(一般标准是至少80%的氨基酸残基的平均3D-1D 值>= 0.2),所以该模型的三维结构不兼容,需要进一步优化。

- ERRAT(分析不同原子类型之间非键合相互作用的统计数据,并绘制误差函数值与9残基滑动窗口位置的关系曲线。)
不同类型的原子在蛋白质中相对于彼此非随机分布。模型构建中的错误导致不同原子类型的更随机分布,这可以通过统计方法与正确分布区分开。
原子分为三类:碳(C),氮(N)和氧(O)。这导致成对非共价键合相互作用(CC,CN,CO,NN,NO和OO)的六种不同组合。二次误差函数用于表征96个可靠蛋白质结构数据库中九个残基滑动窗口的成对相互作用集。然后可以通过分析来自每个窗口的非键合相互作用的模式来鉴定被错误构造的候选蛋白质结构的区域。
Quality Factor: A: 24.2424

(大片红色区域,模型在非键相互作用方面非常不合格)
- WHATCHECK :立体化学
该结构模型存在异常短的原子间距离问题;骨架构造z值非常低的问题;平均B因子错误;HIS, ASN, GLN侧链翻转问题;
【参考链接】
[1] Swiss model建模:https://www.swissmodel.expasy.org/
[2]单模板建模:https://salilab.org/modeller/tutorial/basic.html
[3]多模板建模:https://salilab.org/modeller/tutorial/advanced.html
[4] pdb数据库:http://www.rcsb.org/
[5] uniprot数据库:https://www.uniprot.org/
[6] Molprobity模型评估平台: http://molprobity.biochem.duke.edu/
[7] SAVES模型评估平台:http://servicesn.mbi.ucla.edu/SAVES/