一日千里,追风逐月 | 深势科技发布极致加速版分子对接引擎Uni-Docking


近日,在先后推出Uni-Fold、Uni-FEP、Uni-EM 、Uni-Mol等“Uni-”系列产品后,深势科技继续“追风逐月”,发布GPU极致加速版分子对接引擎Uni-Docking,在NVIDIA V100 GPU上实现了相比单CPU核心超1000倍的加速。研发团队使用Uni-Docking以KRAS G12D为测试靶点,对Enamine Diverse Real类药数据库3820万分子开展了分层高通量虚拟筛选,仅花费11.3小时,助力药物设计人员圆梦“一夜筛遍千万级分子库的高通量虚拟筛选”工作。相关成果以《Uni-Dock: A GPU-Accelerated Docking Program Enables Ultra-Large Virtual Screening》为题[1],发布在ChemRxiv上(点击文末“阅读原文”查看)。

Uni-Docking完善高通量虚拟
筛选的“阿喀琉斯之踵”
发现与目标靶点具有良好结合能力的命中物分子是药物设计早期最重要的目标。虚拟筛选由于具有通量大、速度快、富集能力好的特点,成为发现命中物最重要的手段之一。而其中,分子对接通过计算模拟实现对候选分子与目标靶点的结合活性进行快速评估,表现出了可靠的阳性分子富集能力,被广泛应用于虚拟筛选的过程中[2]。
据估计,潜在的类药性分子化学空间及分子数量已经远远超出已有的早期发现化合物数据库,并随着分子量变大而呈指数上涨[3]。超高通量虚拟筛选,不仅可以帮助药物设计人员在早期筛选中,扩展分子库的化学多样性,还可以同时提升筛选命中化合物的质量[4]。然而,目前主流的分子对接软件计算速度上受到各种因素限制,筛选速度仍然非常有限。同时,受限于有限的计算资源,药物研发机构往往只能对几十万分子开展分子对接,较小的分子数据集规模使得发现优秀命中物的概率更低。另一方面,面对诸如Covid-19新冠肺炎疫情、猴痘病毒感染人体等紧急公共卫生事件,人类对分子对接计算速度提出了更高的要求,期望在最短时间内快速完成大型数据库的超高通量虚拟筛选。基于分子对接的虚拟筛选在待筛库的通量大小和计算速度层面上亟待取得质的突破。
为了满足大型数据库的快速高通量虚拟筛选需求,深势科技团队发布了GPU极致加速的分子对接引擎Uni-Docking,实现了相比单CPU核心超1000倍的加速,同时支持vina、vinardo和ad4打分函数,在速度上超过了现有的GPU加速工作如AutoDock-GPU、Vina-GPU并保持了与Vina持平的精度。研发人员使用Uni-Docking在KRAS G12D靶点上对Enamine Diverse Real类药数据库3820万分层高通量虚拟筛选,使用带有100张NVIDIA V100显卡的计算集群,仅花费11.3小时,平均速度超过3.7万次分子对接/卡时。

Uni-Docking技术实现
Uni-Docking从GPU的大规模并行能力入手,首次实现了多配体、多构象同时对接,对GPU异构加速体系的进行了数据结构与计算逻辑上的深入调优。
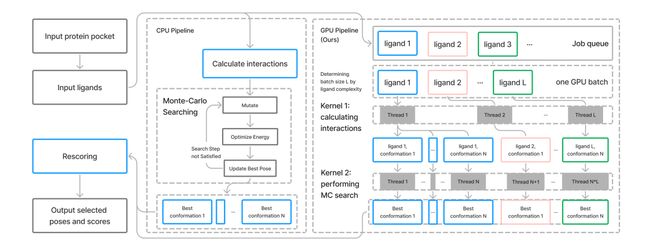
图1: AutoDock Vina 1.2(左)和Uni-Docking(右)的算法架构比较
Uni-Docking采用了和AutoDock Vina 1.2(Vina)相同的构象搜索策略:每个搜索线程对应一个配体构象,用蒙特卡洛方法(Monte Carlo method)全局搜索可旋转键键角、朝向和位置的组合,并在搜索的每一步求能量和力、使用BFGS算法进行梯度下降局部优化,以获取局部能量最低的分子构象。
Uni-Docking实现了多构象并行搜索。通过增加同一个配体的搜索线程数,增加初始构象多样性,实现构象空间的充分覆盖;同时减小每个搜索线程的Monte Carlo迭代步数,通过搜索线程数和迭代步数的调整,实现构象空间的高效搜索。(阶段一)
同时,Uni-Docking实现了多配体并行搜索。在每次GPU调用中同时对数百个配体开展分子对接,并基于显存空间动态分配并行配体数,来充分利用GPU的并行能力,显著缩短了单个配体的平均计算时间。(阶段二)
另外,Uni-Docking通过计算逻辑和数据结构的优化,降低了CPU-GPU之间的通信开销,通过合理的精度优化节省了GPU的显存空间,并通过异步机制合理分配CPU读写文件和GPU计算模拟的时间,来实现Uni-Docking分子对接软件的极致加速。(阶段三)

图2: Uni-Docking三个优化阶段相比AutoDock Vina(exhaustiveness=32)在保持精度持平的情况下,实现120倍、925倍和1627倍的速度提升
我们以AutoDock Vina 1.2在DUD-E中8个靶点的富集能力的表现作为精度衡量标准(相关参数设置为:exhaustiveness=32、Vina打分函数、半柔性对接),实验证明Uni-Docking的三个优化阶段均保持了可比较的计算精度。相比于AutoDock Vina(使用单个Intel® Xeon® Platinum 8269CY〔Cascade Lake〕2.5 GHz CPU核心)对接,Uni-Docking(使用NVIDIA V100 32G GPU)实现了1627倍的对接加速比。

Uni-Docking性能表现
分子对接软件在不同场景下的应用对精度和速度有不同的需求。例如,在大规模数据库的虚拟筛选中通常需要高效率,而在分子结合模式预测中则需要高精度。Uni-Docking提供了三种搜索模式来满足速度和精度的不同需求。
我们使用含有285个蛋白-配体复合物的CASF-2016数据集来评估Uni-Docking预测蛋白-配体结合构象的能力,使用含有102个靶点的DUD-E数据集来评估Uni-Docking富集阳性分子的能力,采用vina打分函数进行实验。另外,我们在DUD-E中的8个靶点上开展了不同打分函数在不同搜索模式下的平均对接时间计算。以上实验使用NVIDIA V100 GPU开展。蛋白残基设置为刚性,小分子设置为柔性。
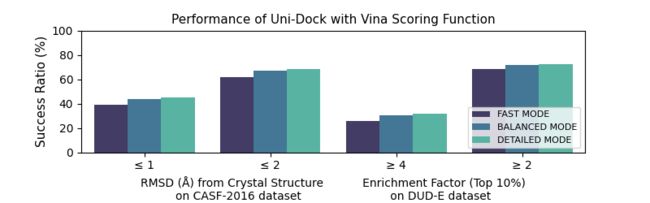
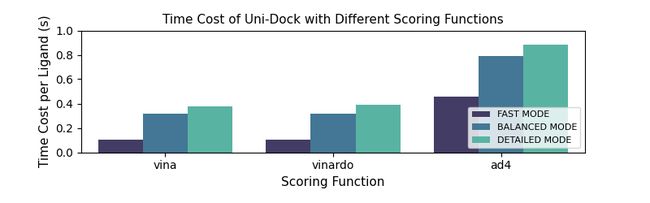
图3: Uni-Docking三种计算模式的计算精度和速度表现
从实验结果可以看到,Uni-Docking Fast模式计算速度快,对接每个配体的平均时间最低仅为0.10s,且保持了良好的阳性分子富集能力,非常适合于特大型数据库的虚拟筛选。Uni-Docking Detailed模式计算精度高,无论在阳性分子富集能力或是结合构象预测上都具有良好的表现。而Uni-Docking Balanced模式在速度和精度上表现较为平均,适用于中等规模数据库的快速准确评估。
另外,Uni-Docking在使用vina和vinardo打分函数时,三种计算模式对应的平均对接时间分别为Fast模式0.10s、Balanced模式0.31s、Detailed模式0.38s;使用ad4打分函数速度分别为Fast模式0.46s、Balanced模式0.79s、Detailed模式0.89s。这样的计算速度意味着极大分子数据库的虚拟筛选能以合理的时间和成本开展。

Uni-Docking筛选KRAS G12D
靶点潜在苗头化合物
我们使用Uni-Docking在KRAS G12D靶点上对Enamine Diverse Real类药数据库3820万分子开展了虚拟筛选。为兼顾速度和精度,我们采用了分层分子对接的方法。首先使用Uni-Docking Fast模式对接了REAL数据集的约3820万个配体。然后,将得分排名前10%的配体(约382万个),使用Uni-Docking Balanced模式进行对接。最后,将得分排名前10%的分子(约38.2万个),使用Uni-Docking Detailed模式进行对接,得到最终结果。

图4: 在Enamine Diverse REAL数据集上针对KRAS G12D靶点进行分层虚拟筛选的时间消耗
我们使用包含100张NVIDIA V100 GPU的GPU集群开展了上述的虚拟对接流程。在11.27小时内完成了上述的虚拟筛选流程。三步不同层级的分子对接分别花费了7.88小时、2.52小时和0.87小时。

总结与未来发展
深势科技Uni-Docking分子对接引擎的发布将基千万级分子数据库的虚拟筛选带入了可及、可用、可靠的“三可”实用时代。Uni-Docking极致的分子对接速度,意味着药物研发项目在药物研发早期能以可接受的成本探索更大的化学空间,有更多的机会发现优良命中物分子,为新药研发提供了更多可能性和更好的基础。同时,作为药物研发的基础工具,Uni-Docking给例如打分函数调优、Uni-Mol数据标注等应用铺平了道路。深势科技将基于Uni-Docking分子对接引擎持续发展,提出更多实用的新药研发工具和策略。
虽然目前Uni-Docking将千万级分子数据库的虚拟筛选带入了实用时代,然而对于十亿级别乃至更大分子空间的虚拟筛选,单一的Uni-Docking引擎并不是最优选择。未来,我们将Uni-Docking与三维分子预训练模型Uni-Mol[5]进行深度融合,为10亿分子数量级以上的超高通量虚拟筛选提供解决方案,敬请关注!
Uni-Docking将于近期完成在Hermite™平台的对接和工程化部署,若您想抢先体验,请扫描文末Hermite™二维码。
参考文献
[1] Yu, Y., Cai C., Zhu Z., Zheng H., 2022. Uni-Dock: A GPU-Accelerated Docking Program Enables Ultra-Large Virtual Screening. DOI: 10.26434/chemrxiv-2022-5t5ts.
[2] Prieto-Martínez FD, Arciniega M, Medina-Franco JL. Molecular docking: current advances and challenges. TIP. Revista especializada en ciencias químico-biológicas. 2018;21.
[3] Lyu, J., Wang, S., Balius, T.E., Singh, I., Levit, A., Moroz, Y.S., O’Meara, M.J., Che, T., Algaa, E., Tolmachova, K. and Tolmachev, A.A., 2019. Ultra-large library docking for discovering new chemotypes. Nature, 566(7743), pp.224-229.
[4] Gorgulla, C., Boeszoermenyi, A., Wang, Z.F., Fischer, P.D., Coote, P.W., Padmanabha Das, K.M., Malets, Y.S., Radchenko, D.S., Moroz, Y.S., Scott, D.A. and Fackeldey, K., 2020. An open-source drug discovery platform enables ultra-large virtual screens. Nature, 580(7805), pp.663-668.
[5] Zhou, G., Gao, Z., Ding, Q., Zheng, H., Xu, H., Wei, Z., Zhang, L. and Ke, G., 2022. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. DOI: 10.26434/chemrxiv-2022-jjm0j.
上下滑动查看更多
关于Hermite™
Hermite™是深势科技打造的基于人工智能、物理建模和高性能计算的新一代药物计算设计平台,致力于为药物研发工作者提供一站式解决方案,满足多种场景的药物研发需求。Hermite™以网页应用的形式,提供多种药物设计功能,帮助药物设计人员完成从靶点结构解析、预测与精修,到苗头化合物筛选,再到先导化合物优化和性质预测的一体化工作。Hermite™提供友好的可视化交互界面、功能实时更新,同时支持本地和云上的私有化部署。
了解和试用Hermite™,请扫码添加平台官方微信,加官方交流群:

现在,在「知乎」也能找到我们了
进入知乎首页搜索「PaperWeekly」
点击「关注」订阅我们的专栏吧
·
