#TCGA系列#利用R处理多个文件夹下的miRNA表达数据
- 当我们TCGA官方下载数据miRNA表达数据时,这些数据大多都位于多个文件夹下,而且有类似文件容易混淆。

- 说实话,R处理文件并没有perl好用,但并不代表不能处理
f1 <- list.files(pattern="isoforms.quantification",recursive=T)
f2 <- list.files(pattern="isoforms.quantification.txt.parcel",recursive=T)
f3 <- setdiff(f1,f2);#取两者的差集
f4 <- strsplit(f3,"/")
f5 <- data.frame(matrix(unlist(f4), nrow=491, byrow=T))
f6 <- as.matrix(f5)
f7 <- f6[,3]
结果展示,最终把文件名取出



- 取出样本后,就该提取数据合为矩阵
#取某个样本,求提取YES后剩的最终行数
da1 <- read.table(f3[1],header = T,stringsAsFactors = F)
da2 <- da1[da1$cross.mapped=="N",]
da3 <- aggregate(read_count~miRNA_region,data=da2,sum)
da <- data.frame(da3$miRNA_region)
colnames(da) <- "miRNA_region"
#利用循环把所有样本的RPM值合为矩阵
for(i in 1:length(f3)){
a <- read.table(f3[i],header = T,stringsAsFactors = F)
b <- a[a$cross.mapped=="N",]
RPM <- aggregate(read_count~miRNA_region,data = b,sum)
RPM$read_count <- (RPM$read_count*10^6)/sum(RPM$read_count)
da <- merge(da,RPM,by="miRNA_region")
}
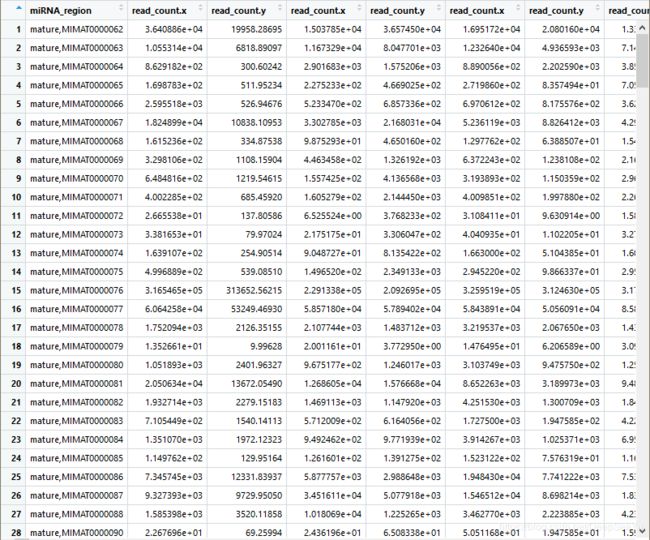
- 修改列名为样本编号
#利用json包给RPM值矩阵改列名
library(rjson)
meta <- jsonlite::fromJSON("metadata.json")
f8 <- meta[,c(3,12)]#提取文件名和样本编号
f9 <- f8[,2]
f10 <- data.frame(matrix(unlist(f9), nrow=2203, byrow=T))
f11 <- cbind(f8[1],f10[3])#合并文件名和样本编号
f12 <- as.data.frame(f7)
colnames(f12) <- "file_name"
f13 <- merge(f12,f11,by="file_name",sort = F)#利用文件名取交集
f14 <- as.character(f13[,2])
colnames(da) <- c("miRNA_region",f14)
write.table(da,file="express.txt",sep="\t",quote=F,row.names=F)
- 数据预处理
#数据预处理
xname <- colnames(da)
xname <- xname[-1]
#给样本分类
tumor <- grep(".*-01[AB]-.*",xname)
control <- grep(".*-11A-.*",xname)
data_control <- da[,c(1,control+1)]
data_tumor <- da[,c(1,tumor+1)]
#一行有超过30%的0时,去除该行
num1 <- apply(data_control[-1,],1,function(x){sum(x==0)})
data_con <- data_control[num1<133,]
num2 <- apply(data_tumor[-1,],1,function(x){sum(x==0)})
data_tum <- data_tumor[num2<14,]
data_all <- merge(data_tum,data_con,by="miRNA_region")
#将矩阵中剩余的0改为0.05
data_all[data_all==0] <- 0.05
write.table(data_all,file="express.txt",sep="\t",quote=F,row.names=F)

注:全部代码如下
setwd("C:\\Users\\Administrator\\Documents\\miRNA")
#读取多个文件夹下的文件
f1 <- list.files(pattern="isoforms.quantification",recursive=T)
f2 <- list.files(pattern="isoforms.quantification.txt.parcel",recursive=T)
f3 <- setdiff(f1,f2);#取两者的差集
f4 <- strsplit(f3,"/")
f5 <- data.frame(matrix(unlist(f4), nrow=491, byrow=T))
f6 <- as.matrix(f5)
f7 <- f6[,3]
#取某个样本,求提取YES后剩的最终行数
da1 <- read.table(f3[1],header = T,stringsAsFactors = F)
da2 <- da1[da1$cross.mapped=="N",]
da3 <- aggregate(read_count~miRNA_region,data=da2,sum)
da <- data.frame(da3$miRNA_region)
colnames(da) <- "miRNA_region"
#利用循环把所有样本的RPM值合为矩阵
for(i in 1:length(f3)){
a <- read.table(f3[i],header = T,stringsAsFactors = F)
b <- a[a$cross.mapped=="N",]
RPM <- aggregate(read_count~miRNA_region,data = b,sum)
RPM$read_count <- (RPM$read_count*10^6)/sum(RPM$read_count)
da <- merge(da,RPM,by="miRNA_region")
}
#利用json包给RPM值矩阵改列名
library(rjson)
meta <- jsonlite::fromJSON("metadata.json")
f8 <- meta[,c(3,12)]#提取文件名和样本编号
f9 <- f8[,2]
f10 <- data.frame(matrix(unlist(f9), nrow=2203, byrow=T))
f11 <- cbind(f8[1],f10[3])#合并文件名和样本编号
f12 <- as.data.frame(f7)
colnames(f12) <- "file_name"
f13 <- merge(f12,f11,by="file_name",sort = F)#利用文件名取交集
f14 <- as.character(f13[,2])
colnames(da) <- c("miRNA_region",f14)
write.table(da,file="express.txt",sep="\t",quote=F,row.names=F)
#数据预处理
xname <- colnames(da)
xname <- xname[-1]
#给样本分类
tumor <- grep(".*-01[AB]-.*",xname)
control <- grep(".*-11A-.*",xname)
data_control <- da[,c(1,control+1)]
data_tumor <- da[,c(1,tumor+1)]
#一行有超过30%的0时,去除该行
num1 <- apply(data_control[-1,],1,function(x){sum(x==0)})
data_con <- data_control[num1<133,]
num2 <- apply(data_tumor[-1,],1,function(x){sum(x==0)})
data_tum <- data_tumor[num2<14,]
data_all <- merge(data_tum,data_con,by="miRNA_region")
#将矩阵中剩余的0改为0.05
data_all[data_all==0] <- 0.05
write.table(data_all,file="express.txt",sep="\t",quote=F,row.names=F)