单细胞计数矩阵是如何生成的?(二)
导读
本文将介绍scRNA-seq的表达矩阵是如何生成。
1. 文库制备
根据所使用的文库制备方法,RNA 序列(也称为读数或标签)将来自转录本(10X Genomics、CEL-seq2、Drop-seq)的 3' 末端(或 5' 末端) , inDrops) 或来自全长转录本 (Smart-seq)。

下面列出了这些方法的以下优点:
-
3’ (or 5’)-end sequencing(3' 端测序):- 通过使用区分生物复制品和扩增 (
PCR) 复制品的独特分子标识符进行更准确的量化 - 测序的细胞数量多,可以更好地识别细胞类型
- 成本更便宜
- 最佳结果大于10,000 个细胞
- 通过使用区分生物复制品和扩增 (
-
Full length sequencing(全长测序):- 检测异构体水平的表达差异
- 鉴定等位基因特异性表达差异
- 对少量细胞进行更深入的测序
- 最适合细胞数量少的样品
3' 端测序与全长测序需要执行许多相同的分析步骤,但 3' 越来越受欢迎,并且在分析中包含更多步骤。因此,将详细分析来自 3' 协议的数据,重点是基于液滴的方法(inDrops、Drop-seq、10X Genomics)。
2. 3’-end
对于 scRNA-seq 数据的分析,了解每个读数中存在哪些信息以及如何在分析中使用它是有帮助的。
对于 3' 端测序方法,源自同一转录本的不同分子的读数将仅源自转录本的 3' 端,因此具有相同序列的可能性很高。然而,文库制备过程中的 PCR 步骤也可能产生重复读取。为了确定读数是生物扩增还是技术扩增,这些方法使用唯一的分子标识符或 UMI。
-
映射到相同转录本的不同
UMI的读取来自不同的分子,并且是生物学重复,每个读取都应该被计算在内。 -
具有相同
UMI的读取来自相同的分子并且是技术重复,应该计为单个读取。 -
在下图中,
ACTB的读取应计为单次读取,而ARL1的读取应分别计数。
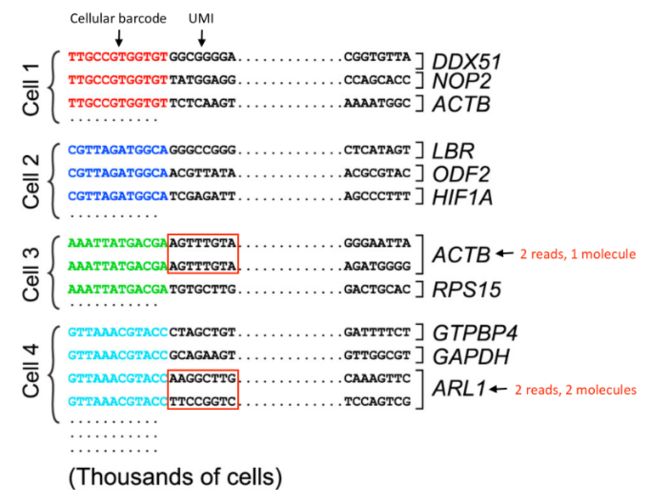
所以需要检查 UMI,无论采用哪种液滴方法,在细胞水平上进行正确量化都需要以下内容:
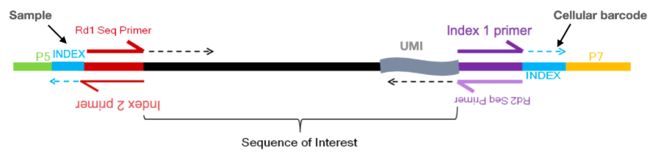
-
Sample index:确定读取来自哪个样本。在文库准备期间添加,需要记录。 -
Cellular barcode:确定读取来自哪个单元格,每种文库制备方法都有一个在文库制备过程中使用的细胞条形码。 -
Unique molecular identifier (UMI):确定读取来自哪个转录分子,UMI将用于折叠 PCR 重复。 -
Sequencing read1:Read1 序列 -
Sequencing read2:Read2 序列
3. 流程
scRNA-seq方法将确定如何从测序读数中解析条形码和 UMI。因此,尽管一些具体步骤会略有不同,但无论采用何种方法,总体工作流程通常都会遵循相同的步骤。一般工作流程如下所示:
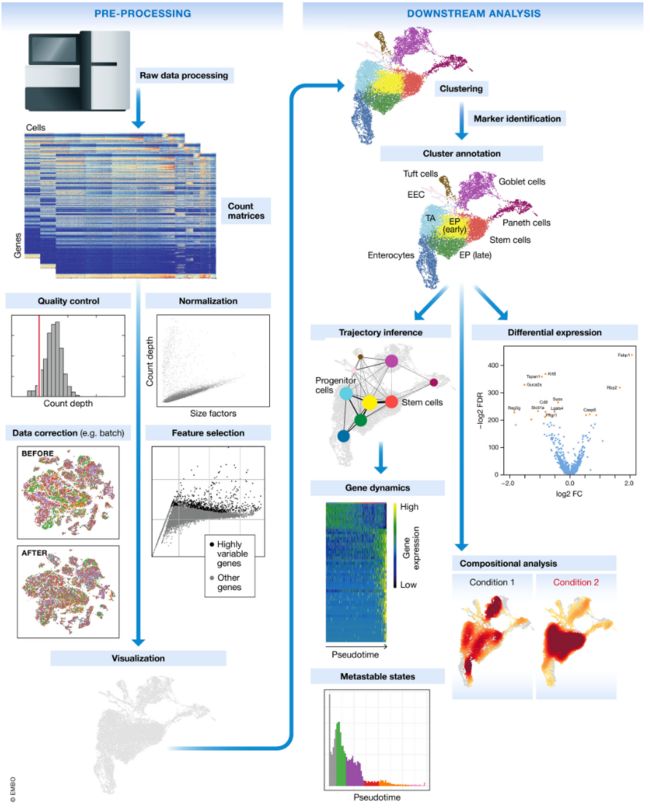 单细胞工作流程
单细胞工作流程
工作流程的步骤是:
- 计数矩阵的生成:
formating reads, demultiplexing samples, mapping and quantification - 原始计数矩阵的质控:过滤劣质细胞
- 聚类:基于转录活性的相似性对细胞进行聚类(细胞类型 类似于 不同的
clusters) -
marker鉴定和簇注释:识别每个簇的marker并注释已知的细胞类型簇 - 下游其他分析
无论进行何种分析,基于每个条件的单个样本得出的关于总体的结论都是不可信的。仍然需要生物重复!也就是说,如果您想得出与总体相对应的结论,请做生物学重复。
4. 计数矩阵
首先讨论此工作流程的第一部分,即从原始测序数据生成计数矩阵。将重点关注基于液滴的方法使用的 3' 端测序,例如 inDrops、10X Genomics 和 Drop-seq。
测序后,要么将原始测序数据输出为 BCL 或 FASTQ 格式,要么生成计数矩阵。如果读取是 BCL 格式,那么需要转换为 FASTQ 格式。 bcl2fastq 工具可以轻松执行此转换。
对于许多 scRNA-seq 方法,从原始测序数据生成计数矩阵经历的步骤类似。
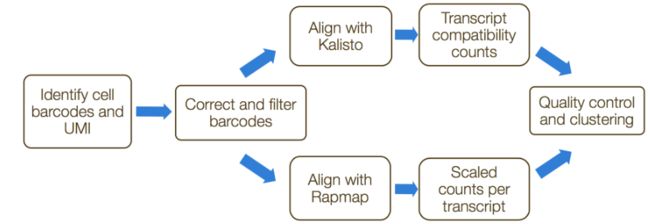
alevin[1] 是一个命令行工具,用于估计 scRNA-seq 数据的表达,其中转录物的 3' 末端被测序。umi-tools[2] 和 zUMI[3] 是可以执行这些过程的附件。这些工具结合了 UMI 以纠正假阳性。此过程中的步骤包括:
- 格式化读取和过滤嘈杂的
cellular barcodes - 样本拆分
-
Mapping到转录组 - 根据
UMI进行定量
如果使用 10X Genomics 文库制备方法,则 Cell Ranger 管道包含上述所有步骤。
5. 数据解析
使用 FASTQ 文件解析cell barcodes, UMIs, 和 sample barcodes。对于基于液滴的方法,由于以下原因,许多细胞条形码将匹配少量读取(< 1000 读取):
- 从垂死的细胞中包裹自由漂浮的 RNA
- 表达少量基因的细胞(红细胞等)
- 由于未知原因死亡的细胞
在读取结果之前,需要从序列数据中过滤掉这些多余的条形码。为了进行这种过滤,提取并保存每个细胞的cellular barcode和molecular barcode。例如,如果使用umis工具,信息将添加到每次读取的header,格式如下:
@HWI-ST808:130:H0B8YADXX:1:1101:2088:2222:CELL_GGTCCA:UMI_CCCT
AGGAAGATGGAGGAGAGAAGGCGGTGAAAGAGACCTGTAAAAAGCCACCGN
+
@@@DDBD>=AFCF+#
文库制备方法中使用的细胞条形码(cellular barcodes)是已知的,未知条形码将被丢弃,同时允许与已知细胞条形码的数量不匹配。
6. 数据拆分
如果对多个样本进行测序,则下一步是对样本进行拆分。这个过是由zUMIs完成的。需要解析读取以确定与每个单元格相关的样本条形码(sample barcode)。
7. 比对
为了确定reads来自哪个基因,使用传统 STAR 或Kallisto/RapMap进行align(对齐)。
8. 定量
重复的 UMI 被去除,只有唯一的 UMI 使用 Kallisto 或 featureCounts 等工具进行定量。结果输出是一个细胞的基因计数矩阵:
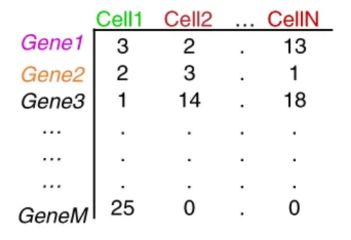 计数矩阵
计数矩阵
矩阵中的每个值表示来自相应基因的单元格中的读取数。使用计数矩阵,可以探索和过滤数据,只保留高质量的单元格。
参考资料
[1]alevin: https://salmon.readthedocs.io/en/latest/alevin.html
[2]umi-tools: https://hbctraining.github.io/scRNA-seq_online/lessons/02_SC_generation_of_count_matrix.html
[3]zUMI: https://github.com/sdparekh/zUMIs
本文由 mdnice 多平台发布