Nanopore sequencing and assembly of a human genome with ultra-long reads
Nanopore sequencing and assembly of a human genome with ultra-long reads
- Miten Jain,
- Sergey Koren,
- […]
- Matthew Loose
Nature Biotechnology volume 36, pages338–345(2018)Cite this article
-
55k Accesses
-
433 Citations
-
1509 Altmetric
-
Metricsdetails
Abstract
We report the sequencing and assembly of a reference genome for the human GM12878 Utah/Ceph cell line using the MinION (Oxford Nanopore Technologies) nanopore sequencer. 91.2 Gb of sequence data, representing ∼30× theoretical coverage, were produced. Reference-based alignment enabled detection of large structural variants and epigenetic modifications. De novo assembly of nanopore reads alone yielded a contiguous assembly (NG50 ∼3 Mb). We developed a protocol to generate ultra-long reads (N50 > 100 kb, read lengths up to 882 kb). Incorporating an additional 5× coverage of these ultra-long reads more than doubled the assembly contiguity (NG50 ∼6.4 Mb). The final assembled genome was 2,867 million bases in size, covering 85.8% of the reference. Assembly accuracy, after incorporating complementary short-read sequencing data, exceeded 99.8%. Ultra-long reads enabled assembly and phasing of the 4-Mb major histocompatibility complex (MHC) locus in its entirety, measurement of telomere repeat length, and closure of gaps in the reference human genome assembly GRCh38.
Main
The human genome is used as a yardstick to assess performance of DNA sequencing instruments1,2,3,4,5. Despite improvements in sequencing technology, assembling human genomes with high accuracy and completeness remains challenging. This is due to size (∼3.1 Gb), heterozygosity, regions of GC% bias, diverse repeat families, and segmental duplications (up to 1.7 Mbp in size) that make up at least 50% of the genome6. Even more challenging are the pericentromeric, centromeric, and acrocentric short arms of chromosomes, which contain satellite DNA and tandem repeats of 3–10 Mb in length7,8. Repetitive structures pose challenges for de novo assembly using “short read” sequencing technologies, such as Illumina's. Such data, while enabling highly accurate genotyping in non-repetitive regions, do not provide contiguous de novo assemblies. This limits the ability to reconstruct repetitive sequences, detect complex structural variation, and fully characterize the human genome.
Single-molecule sequencers, such as Pacific Biosciences' (PacBio), can produce read lengths of 10 kb or more, which makes de novo human genome assembly more tractable9. However, single-molecule sequencing reads have significantly higher error rates compared with Illumina sequencing. This has necessitated development of de novo assembly algorithms and the use of long noisy data in conjunction with accurate short reads to produce high-quality reference genomes10. In May 2014, the MinION nanopore sequencer was made available to early-access users11. Initially, the MinION nanopore sequencer was used to sequence and assemble microbial genomes or PCR products12,13,14 because the output was limited to 500 Mb to 2 Gb of sequenced bases. More recently, assemblies of eukaryotic genomes including yeasts, fungi, and Caenorhabditis elegans have been reported15,16,17.
Recent improvements to the protein pore (a laboratory-evolved Escherichia coli CsgG mutant named R9.4), library preparation techniques (1D ligation and 1D rapid), sequencing speed (450 bases/s), and control software have increased throughput, so we hypothesized that whole-genome sequencing (WGS) of a human genome might be feasible using only a MinION nanopore sequencer17,18,19.
We report sequencing and assembly of a reference human genome for GM12878 from the Utah/CEPH pedigree, using MinION R9.4 1D chemistry, including ultra-long reads up to 882 kb in length. GM12878 has been sequenced on a wide variety of platforms, and has well-validated variation call sets, which enabled us to benchmark our results20.
Results
Sequencing data set
Five laboratories collaborated to sequence DNA from the GM12878 human cell line. DNA was sequenced directly (avoiding PCR), thus preserving epigenetic modifications such as DNA methylation. 39 MinION flow cells generated 14,183,584 base-called reads containing 91,240,120,433 bases with a read N50 (the read length such that reads of this length or greater sum to at least half the total bases) of 10,589 bp (Supplementary Tables 1–4). Ultra-long reads were produced using 14 additional flow cells. Read lengths were longer when the input DNA was freshly extracted from cells compared with using Coriell-supplied DNA (Fig. 1a). Average yield per flow cell (2.3 Gb) was unrelated to DNA preparation methods (Fig. 1b). 94.15% of reads had at least one alignment to the human reference (GRCh38) and 74.49% had a single alignment over 90% of their length. Median coverage depth was 26-fold, and 96.95% (3.01/3.10 Gbp) of bases of the reference were covered by at least one read (Fig. 1c). The median identity of reads was 84.06% (82.73% mean, 5.37% s.d.). No length bias was observed in the error rate with the MinION (Fig. 1d).
Figure 1: Summary of data set.

(a) Read length N50s by flow cell, colored by sequencing center. Cells: DNA extracted directly from cell culture. DNA: pre-extracted DNA purchased from Coriell. UoB, Univ. Birmingham; UEA, Univ. East Anglia; UoN, Univ. Nottingham; UBC, Univ. British Columbia; UCSC, Univ. California, Santa Cruz. (b) Total yield per flow cell grouped as in a. (c) Coverage (black line) of GRCh38 reference compared to a Poisson distribution. The depth of coverage of each reference position was tabulated using samtools depth and compared with a Poisson distribution with lambda = 27.4 (dashed red line). (d) Alignment identity compared to alignment length. No length bias was observed, with long alignments having the same identity as short ones. (e) Correlation between 5-mer counts in reads compared to expected counts in the chromosome 20 reference. (f) Chromosome 20 homopolymer length versus median homopolymer base-call length measured from individual Illumina and nanopore reads (Scrappie and Metrichor). Metrichor fails to produce homopolymer runs longer than ∼5 bp. Scrappie shows better correlation for longer homopolymer runs, but tends to overcall short homopolymers (between 5 and 15 bp) and undercall long homopolymers (>15 bp). Plot noise for longer homopolymers is due to fewer samples available at that length.
Full size image
Base-caller evaluation
The base-calling algorithm used to decode raw ionic current signal can affect sequence calls. To analyze this effect we used reads mapping to chromosome 20 and compared base-calling with Metrichor (an LSTM-RNN base-caller) and Scrappie, an open-source transducer neural network (Online Methods). Of note, we observed that a fraction of the Scrappie output (4.7% reads, 14% bases) was composed of low-complexity sequence (Supplementary Fig. 1), which we removed before downstream analysis.
To assess read accuracy we realigned reads from each base-caller using a trained alignment model21. Alignments generated by the Burrows–Wheeler Aligner Maximal Exact Matches (BWA-MEM) were chained such that each read had at most one maximal alignment to the reference sequence (scored by length). The chained alignments were used to derive the maximum likelihood estimate of alignment model parameters22, and the trained model used to realign the reads. The median identity after realignment for Metrichor was 82.43% and for Scrappie, 86.05%. We observed a purine-to-purine substitution bias in chained alignments where the model was not used (Supplementary Fig. 2). The alignments produced by the trained model showed an improved substitution error rate, decreasing the overall transversion rate, but transition errors remained dominant.
To measure potential bias at the k-mer level, we compared counts of 5-mers in reads derived from chromosome 20. In Metrichor reads, the most underrepresented 5-mers were A/T-rich homopolymers. The most overrepresented k-mers were G/C-rich and non-homopolymeric (Supplementary Table 5). By contrast, Scrappie showed no underrepresentation of homopolymeric 5-mers and had a slight overrepresentation of A/T homopolymers. Overall, Scrappie showed the lowest k-mer representation bias (Fig. 1e). The improved homopolymer resolution of Scrappie was confirmed by inspection of chromosome 20 homopolymer calls versus the human reference (Fig. 1f and Supplementary Fig. 3)23. Despite this reduced bias, whole-genome assembly and analyses proceeded with Metrichor reads, since Scrappie was still in early development at the time of writing.
De novo assembly of nanopore reads
We carried out a de novo assembly of the 30× data set with Canu24 (Table 1). This assembly comprised 2,886 contigs with an NG50 contig size of 3 Mbp (NG50, the longest contig such that contigs of this length or greater sum to at least half the haploid genome size). The identity to GRCh38 was estimated as 95.20%. Canu was fourfold slower on the Nanopore data compared to a random subset of equivalent coverage of PacBio data requiring ∼62K CPU hours. The time taken by Canu increased when the input was nanopore sequence reads because of systematic error in the raw sequencing data leading to reduced accuracy of the Canu-corrected reads, an intermediate output of the assembler. Corrected PacBio reads are typically >99% identical to the reference; our reads averaged 92% identity to the reference after correction (Supplementary Fig. 1b).
Table 1 Summary of assembly statistics
Full size table
We aligned assembled contigs to the GRCh38 reference and found that our assembly was in agreement with previous GM12878 assemblies (Supplementary Fig. 4)25. The number of structural differences (899) that we identified between GM12878 and GRCh38 was similar to that of a previously published PacBio assembly of GM12878 (692) and of other human genome assemblies5,24, but with a higher than expected number of deletions, due to consistent truncation of homopolymer and low-complexity regions (Supplementary Fig. 5 and Supplementary Table 6). Consensus identity of our assembly with GRCh38 was estimated to be 95.20% (Table 1). However, GRCh38 is a composite of multiple human haplotypes, so this is a lower bound on accuracy. Comparisons with independent Illumina data from GM12878 yielded a higher accuracy estimate of 95.74%.
Despite the low consensus accuracy, contiguity was good. For example, the assembly included a single ∼3-Mbp contig that had all class I human leukocyte antigens (HLA) genes from the major histocompatibility complex (MHC) region on chromosome 6, a region notoriously difficult to assemble using short reads. The more repetitive class II HLA gene locus was fragmented but most genes were present in a single contig.
Genome polishing
To improve the accuracy of our assembly we mapped previously generated whole-genome Illumina data (SRA: ERP001229) to each contig using BWA-MEM and corrected errors using Pilon. This improved the estimated accuracy of our assembly to 99.29% versus GRCh8 and 99.88% versus independent GM12878 sequencing (Table 1 and Supplementary Fig. 6)26. This estimate is a lower bound as true heterozygous variants and erroneously mapped sequences decrease identity. Recent PacBio assemblies of mammalian genomes that were assembled de novo and polished with Illumina data exceed 99.95%9,27. Pilon cannot polish regions that have ambiguous short-read mappings, that is, in repeats. We also compared the accuracy of our polished assembly in regions with expected coverage versus those that had low-quality mappings (either lower coverage or higher than expected coverage with low mapping quality) versus GRCh38. When compared to GRCh38, accuracy in well-covered regions increased to 99.32% from the overall accuracy of 99.29%, while the poorly covered regions accuracy dropped to 98.65%.
For further evaluation of our assembly, we carried out comparative annotation before and after polishing (Supplementary Table 7). 58,338 genes (19,436 coding, 96.4% of genes in GENCODE V24, 98.2% of coding genes) were identified representing 179,038 transcripts in the polished assembly. Reflecting the assembly's high contiguity, only 857 (0.1%) of genes were found on two or more contigs.
Alternative approaches to improve assembly accuracy using different base-callers and exploiting the ionic current signal were attempted on a subset of reads from chromosome 20. Assembly consensus improvement using raw output is commonly used when assembling single-molecule data. To quantify the effect of base-calling on the assembly, we reassembled the read sets from Metrichor and Scrappie with the same Canu parameters used for the whole-genome data set. While all assemblies had similar contiguity, using Scrappie reads improved accuracy from 95.74% to 97.80%. Signal-level polishing of Scrappie-assembled reads using nanopolish increased accuracy to 99.44%, and polishing with Illumina data brought the accuracy up to 99.96% (Table 1).
Analysis of sequences not in the assembly
To investigate sequences omitted from the primary genome analysis, we assessed 1,425 contigs filtered from Canu due to low coverage, or contigs that were single reads with many shorter reads within (26 Mbp), or corrected reads not incorporated into contigs (10.4 Gbp). Most sequences represented repeat classes, for example, long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) (Supplementary Fig. 7), observed in similar proportion in the primary assembly, with the exception of satellite DNAs known to be enriched in human centromeric regions. These satellites were enriched 2.93× in the unassembled data and 7.9× in the Canu-filtered contigs. We identified 56 assembled contigs containing centromere repeat sequences specific to each of the 22 autosomes and X chromosome. The largest assembled satellite in these contigs is a 94-kbp tandem repeat specific to centromere 15 (D15Z1, tig00007244).
SNP and SV genotyping
Using SVTyper28 and Platinum Illumina WGS alignments, we genotyped 2,414 GM12878 structural variants (SVs), which were previously identified using LUMPY and validated with PacBio and/or Moleculo reads29. We then genotyped the same SVs using alignments of our nanopore reads from the 30×-coverage data set and a modified version of SVTyper. We measured the concordance of genotypes at each site in the Illumina- and nanopore-derived data, deducing the sensitivity of SV genotyping as a function of nanopore sequencing depth (Fig. 2a). When all 39 flow cells were used, nanopore data recovered 91% of high-confidence SVs with a false-positive rate of 6%. Illumina and nanopore genotypes agreed at 81% of heterozygous sites and 90% of homozygous alternate sites. Genotyping heterozygous SVs using nanopore alignments was limited when homopolymer stretches occur at the breakpoints of these variants (Supplementary Fig. 8a). We determined Illumina, nanopore, and PacBio genotype concordance at a set of 2,192 deletions common to our high-confidence set and a genotyped SV call set derived from PacBio sequencing of GM12878 (refs. 5, 30). PacBio and Illumina genotypes agreed at 94% of heterozygous and 79% of homozygous alternate deletions; nanopore and Illumina genotypes agreed at 90% of heterozygous and 90% of homozygous alternate sites; nanopore and PacBio genotypes agreed at 91% of heterozygous and 76% of homozygous alternate sites. Nearly a quarter (44) of the homozygous alternate sites at which PacBio and Illumina genotypes disagreed overlapped SINEs or LINEs. By manual inspection in the integrative genomics viewer (IGV)31, we observed that sequencing reads were spuriously aligned at these loci and likely drove the discrepancy in predicted genotypes (Supplementary Fig. 8b).
Figure 2: Structural variation and SNP genotyping.
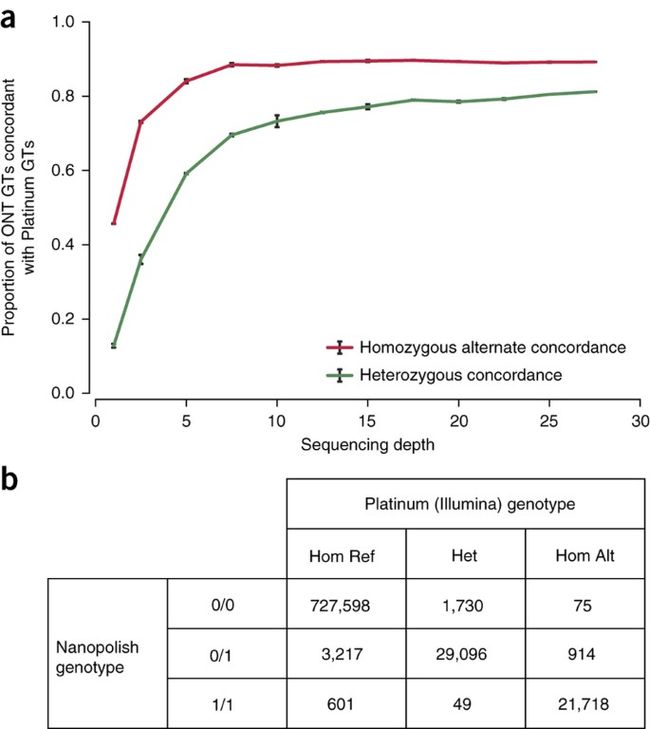
(a) Structural variant genotyping sensitivity using Oxford Nanopore Technologies (ONT) reads. Genotypes (GTs) were inferred for a set of 2,414 SVs using both Oxford Nanopore and Platinum Genomes (Illumina) alignments. Using alignments randomly subsampled to a given sequencing depth (n = 3), sensitivity was calculated as the proportion of ONT-derived genotypes that were concordant with Illumina-derived genotypes. (b) Confusion matrix for genotype-calling evaluation. Each cell contains the number of 1000 Genome sites for a particular nanopolish/platinum genotype combination.
Full size image
We evaluated nanopore data for calling genotypes at known single-nucleotide polymorphisms (SNPs) using the ionic current by calling genotypes at non-singleton SNPs on chromosome 20 from phase 3 of the 1000 Genomes32 and comparing these calls to Illumina Platinum Genome calls (Fig. 2b). 99.16% of genotype calls were correct (778,412 out of 784,998 sites). This result is dominated by the large number of homozygous reference sites. If we assess accuracy by the fraction of correctly called variant sites (heterozygous or homozygous non-reference), the accuracy of our caller is 91.40% (50,814 out of 55,595), with the predominant error being miscalling sites labeled homozygous in the reference as heterozygous (3,217 errors). Genotype accuracy, when only considering sites annotated as variants in the platinum call set, is 94.83% (50,814 correct out of 53,582).
Detection of epigenetic 5-methylcytosine modification
Changes in the ionic current when modified and unmodified bases pass through the MinION nanopores enable detection of epigenetic marks33,34. We used nanopolish and SignalAlign to map 5-methylcytosine at CpG dinucleotides as detected in our sequencing reads against chromosome 20 of the GRCh38 reference35,36. Nanopolish outputs a frequency of reads calling a methylated cytosine, and SignalAlign outputs a marginal probability of methylation summed over reads. We compared the output of both methods to published bisulfite sequencing data from the same DNA region (ENCFF835NTC). Good concordance of our data with the published bisulfite sequencing was observed; the r-values for nanopolish and SignalAlign were 0.895 and 0.779, respectively (Fig. 3 and Supplementary Figs. 9 and 10).
Figure 3: Methylation detection using signal-based methods.

(a) SignalAlign methylation probabilities compared to bisulfite sequencing frequencies at all called sites. (b) Nanopolish methylation frequencies compared to bisulfite sequencing at all called sites. (c) SignalAlign methylation probabilities compared to bisulfite sequencing frequencies at sites covered by at least ten reads in the nanopore and bisulfite data sets; reads were not filtered for quality. (d) Nanopolish methylation frequencies compared to bisulfite sequencing at sites covered by at least ten reads in the nanopore and bisulfite data sets. A minimum log-likelihood threshold of 2.5 was applied to remove ambiguous reads. N = sample size, r = Pearson correlation coefficient.
Full size image
Ultra-long reads improve phasing and assembly contiguity
We modeled the contribution of read length to assembly quality, predicting that ultra-long read data sets (N50 >100 kb) would substantially improve assembly contiguity (Fig. 4a). We developed a method to produce ultra-long reads by saturating the Oxford Nanopore Rapid Kit with high molecular weight DNA. In so doing we generated an additional 5× coverage (Supplementary Fig. 11). Two additional standard protocol flow cells generated a further 2× coverage and were used as controls for software and base-caller versions. The N50 read length of the ultra-long data set was 99.7 kb (Fig. 4b). Reads were impossible to align efficiently at first, because aligner algorithms are optimized for short reads. Further, CIGAR strings generated by ultra-long reads do not fit in the BAM format specification, necessitating the use of SAM or CRAM formats only (https://github.com/samtools/hts-specs/issues/40). Instead, we used GraphMap37 to align ultra-long reads to GRCh38, which took >25K CPU hours (Supplementary Table 8). Software optimized for long reads, including NGM-LR38 and Minimap2 (ref. 39), were faster: Minimap2 took 60 CPU hours. More than 80% of bases were in sequences aligned over 90% of their length with GraphMap and more than 60% with minimap2. Median alignment identity was 81% (83 with minimap2), slightly lower than observed for the control flow cells (83.46%/84.64%) and the original data set (83.11%/84.32%). The longest full-length mapped read in the data set (aligned with GraphMap) was 882 kb, corresponding to a reference span of 993 kb.
Figure 4: Repeat modeling and assembly.

(a) A model of expected NG50 contig size when correctly resolving human repeats of a given length and identity. The y axis shows the expected NG50 contig size when repeats of a certain length (x axis) or sequence identity (colored lines) can be consistently resolved. Nanopore assembly contiguity (GM12878 20×, 30×, 35×) is currently limited by low coverage of long reads and a high error rate, making repeat resolution difficult. These assemblies approximately follow the predicted assembly contiguity. The projected assembly contiguity using 30 × of ultra-long reads (GM12878 30× ultra) exceeds 30 Mbp. A recent assembly of 65 × PacBio P6 data with an NG50 of 26 Mbp is shown for comparison (CHM1 P6). (b) Yield by read length (log10) for ligation, rapid and ultra-long rapid library preparations. (c) Chromosomes plot illustrating the contiguity of the nanopore assembly boosted with ultra-long reads. Contig and alignment boundaries, not cytogenetic bands, are represented by a color switch, so regions of continuous color indicate regions of contiguous sequence. White areas indicate unmapped sequence, usually caused by N's in the reference genome. Regions of interest, including the 12 50+ kb gaps in GRCh38 closed by our assembly as well as the MHC (16 Mbp), are outlined in red.
Full size image
The addition of 5× coverage ultra-long reads more than doubled the previous assembly NG50 to 6.4 Mbp and resolved the MHC locus into a single contig (Fig. 4c). In comparison, a 50× PacBio GM12878 data set with average read length of 4.5 kb assembled with an NG50 contig size of 0.9 Mbp5. Newer PacBio assemblies of a human haploid cell line, with mean read lengths greater than 10 kb, have reached contig NG50s exceeding 20 Mbp at 60× coverage25. We subsampled this data set to a depth equivalent to ours (35×) and assembled, resulting in an NG50 of 5.7 Mbp, with the MHC split into >2 contigs. The PacBio assembly is less contiguous, despite a higher average read length and simplified haploid genome.
In addition to assembling the MHC into a single contig, the ultra-long MinION reads enabled the contiguous MHC to be haplotype phased. Due to the limited depth of nanopore reads, heterozygous SNPs were called using Illumina data and then phased using the ultra-long nanopore reads to generate two pseudo-haplotypes, from which MHC typing was performed using the approach of Dilthey et al.40 (Fig. 5a). Some gaps were introduced during haplotig (contigs with the same haplotype) assembly, owing to low phased-read coverage—for example, HLA-DRB3 was left unassembled on haplotype A—but apart from one HLA-DRB1 allele, sample HLA types wererecovered almost perfectly with an edit distance of 0–1 for true allele versus called allele (Supplementary Table 9). Analysis of parental (GM12891, GM12892) HLA types confirmed the absence of switch errors between the classical HLA typing genes. To our knowledge, this is the first time the MHC has been assembled and phased over its full length in a diploid human genome.
Figure 5: Ultra-long reads, assembly, and telomeres.

(a) A 16-Mbp ultra-long read contig and associated haplotigs are shown spanning the full MHC region. MHC Class I and II regions are annotated along with various HLA genes. Below this contig, the MHC region is enlarged, showing haplotype A and B coverage tracks for the phased nanopore reads. Nanopore reads were aligned back to the polished Canu contig, with colored lines indicating a high fraction of single-nucleotide discrepancies in the read pileups (as displayed by the IGV31 browser). The many disagreements indicate the contig is a mosaic of both haplotypes. The haplotig A and B tracks show the result of assembling each haplotype read set independently. Below this, the MHC class II region is enlarged, with haplotype A and B raw reads aligned to their corresponding, unpolished haplotigs. The few consensus disagreements between raw reads and haplotigs indicate successful partitioning of the reads into haplotypes. (b) An unresolved, 50-kb bridged scaffold gap on Xq24 remains in the GRCh38 assembly (adjacent to scaffolds AC008162.3 and AL670379.17, shown in green). This gap spans a ∼4.6-kb tandem repeat containing cancer/testis gene family 47 (CT47). This gap is closed by assembly (contig: tig00002632) and has eight tandem copies of the repeat, validated by alignment of 100 kb+ ultra-long reads also containing eight copies of the repeat (light blue with read name identifiers). One read has only six repeats, suggesting the tandem repeated units are variable between homologous chromosomes. (c) Ultra-long reads can predict telomere length. Two 100 kb+ reads that map to the subtelomeric region of the chromosome 21 q-arm, each containing 4.9–9.1 kb of the telomeric (TTAGGG_ repeat). (d) Telomere length estimates showing variable lengths between non-homologous chromosomes.
Full size image
Already published single-molecule human genome assemblies contain multiple contigs that span the MHC5,41,42 and phasing has not been attempted. Instead, MHC surveys have focused on homozygous cell lines43.
Ultra-long reads close gaps in the human reference genome
Large (>50 kb) bridged scaffold gaps remain unresolved in the reference human genome assembly (GRCh38). These breaks in the assembly span tandem repeats and/or long tracts of segmental duplications44. Using sequence from our de novo–assembled contigs, we were able to close 12 gaps, each of which was more than 50 kb in the reference genome. We then looked for individual ultra-long reads that spanned gaps, and matched the sequence closure for each region as predicted by the assembly (Supplementary Table 10).
The gap closures enabled us to identify 83,980 bp of previously unknown euchromatic sequence. For example, an unresolved 50-kbp scaffold gap on Xq24 marks the site of a human-specific tandem repeat that contains a cancer/testis gene family, known as CT47 (refs. 45, 46). This entire region is spanned by a single contig in our final assembly (tig00002632). Inspection of this contig using hidden Markov model (HMM) profile modeling of an individual repeat unit containing the CT47A11 gene (GRCh38 chrX:120932333–120938697) suggests that there is an array of eight tandem copies of the CT47 repeat (Fig. 5b). In support of this finding, we identified three ultra-long reads that together traversed the entire tandem array (Fig. 5b); two reads provide evidence for an array of eight repeat copies and one read supports six copies, suggesting heterozygosity.
Telomere repeat lengths
FISH (fluorescent in situ hybridization) estimates and direct cloning of telomeric DNA suggests that telomere repeats (TTAGGG) extend for multiple kilobases at the ends of each chromosome47,48. Using HMM profile modeling of the published telomere tract of repeats (M19947.1), we identified 140 ultra-long reads that contained the TTAGGG tandem repeat (Supplementary Table 11). Sequences next to human telomeres are enriched in intra- and interchromosomal segmental duplications, which makes it difficult to map ultra-long reads directly to the chromosome assemblies. However, we were able to map 17/140 ultra-long reads to specific chromosome subtelomeric regions. We analyzed the mapped regions by identifying the junction or the start of the telomeric array on 17 ultra-long reads, and annotating all TTAGGG-repeat sequences to the end of the read to estimate telomeric repeat length. For example, two reads that only mapped to chromosome 21q indicate that there are 9,108 bp of telomeric repeats. Overall, we found evidence for telomeric arrays that span 2–11 kb within 14 subtelomeric regions for GM12878 (Fig. 5c,d and Supplementary Table 11).
Discussion
We report sequencing and assembly of a human genome with 99.88% accuracy and an NG50 of 6.4 Mb using unamplified DNA and nanopore reads followed by short-read consensus improvement. At 30× coverage we have produced the most contiguous assembly of a human genome to date, using only a single sequencing technology and the Canu assembler23. Consistent with the view that the underlying ionic raw current contains additional information, signal-based polishing14 improved the assembly accuracy to 99.44%. Finally, we report that combining signal-based polishing and short-read (Illumina) correction26 gave an assembly accuracy of 99.96%, which is similar to metrics for other mammalian genomes9.
Here we report that read lengths produced by the MinION nanopore sequencer were dependent on the input fragment length. We found that careful preparation of DNA in solution using classical extraction and purification methods can yield extremely long reads. The longest read lengths were achieved using the transposase-based rapid library kit in conjunction with methods of DNA extraction designed to mitigate shearing. We produced 5× coverage with ultra-long reads, and used this data set to augment our initial assembly. The final 35× coverage assembly has an NG50 of 6.4 Mb. Based on modeling we predict that 30× of ultra-long reads alone would result in an assembly with a contig NG50 in excess of 40 Mb, approaching the contiguity of the current human reference (Fig. 4c). We posit that there may be no intrinsic read-length limit for pore-based sequencers, other than from physical forces that lead to DNA fragmentation in solution. Therefore, there is scope to further improve the read-length results obtained here, perhaps through solid-phase DNA extraction and library preparation techniques, such as agar encasement.
The increased single-molecule read length that we report here, obtained using a MinION nanopore sequencer, enabled us to analyze regions of the human genome that were previously intractable with state-of-the-art sequencing methods. For example, we were able to phase megabase regions of the human genome in single contigs, to more accurately estimate telomere lengths, and to resolve complex repeat regions. Phasing of 4- to 5-Mb scaffolds through the MHC has recently been reported using a combination of sequencing and genealogical data49. However, the resulting assemblies contained multiple gaps of unknown sequences. We phased the entire MHC, and reconstructed both alleles. Development of tools to automate phasing from nanopore assemblies is now needed.
We also wrote custom software/algorithms (poredb) to track the large number of reads, store each read as an individual file, and enable use of cloud-based pipelines for our analyses.
Our proof-of-concept demonstration of human genome sequencing using a MinION nanopore sequencer reveals the potential of this approach, but identifies specific challenges for future projects. Improvements in real-time base-calling are needed to simplify the workflow. More compact and convenient formats for storing raw and base-called data are urgently required, ideally employing a standardized, streaming compatible serialization format such as BAM/CRAM.
With ultra-long reads we found the longest reads exceeded CIGAR string limitations in the BAM format, necessitating the use of SAM or CRAM (https://github.com/samtools/hts-specs/issues/40). And, we were unable to complete an alignment of the ultra-long reads using BWA-MEM, and needed to adopt other algorithms, including GraphMap and NGM-LR, to align the reads. This required large amounts of compute time and RAM37,38,50. Availability of our data set has spurred the development of Minimap2 (ref. 39), and we recommend this long-read aligner for use in aligning ultra-long reads on a standard desktop computer.
Nanopore genotyping accuracy currently lags behind short-read sequencing instruments, due to a limited ability to discriminate between heterozygous and homozygous alleles, which arose from error rate and the depth of coverage in our sequencing data. We found that >99% of SNP calls were correct at homozygous reference sites, dropping to 91.4% at heterozygous and homozygous non-reference sites. Similarly, Nanopore and Illumina SV genotypes agreed at 81% of heterozygous and 90% of homozygous sites. These results highlight a need for structural variant genotyping tools for long, single-molecule sequencing reads. Using 1D2 chemistry (which sequences template and complement strands of the same molecule) or modeling nanopore ionic raw current, perhaps by incorporating training data from modified DNA, could potentially produce increased read accuracy. A complementary approach would be to increase coverage.
In summary, we provide evidence that a portable, biological nanopore sequencer could be used to sequence, assemble, and provisionally analyze structural variants and detect epigenetic marks, in point-of-care human genomics applications in the future.
Methods
Human DNA.
Human genomic DNA from the GM12878 human cell line (CEPH/Utah pedigree) was either purchased from Coriell as DNA (cat. no. NA12878) or extracted from the cultured cell line also purchased from Coriell (cat. no. GM12878). Cell culture was performed using Epstein–Barr virus (EBV)-transformed B lymphocyte culture from the GM12878 cell line in RPMI-1640 media with 2 mM L-glutamine and 15% FBS at 37 °C.
QIAGEN DNA extraction.
DNA was extracted from cells using the QIAamp DNA mini kit (Qiagen). 5 × 106 cells were spun at 300g for 5 min to pellet. The cells were resuspended in 200 μl PBS and DNA was extracted according to the manufacturer's instructions. DNA quality was assessed by running 1 μl on a genomic ScreenTape on the TapeStation 2200 (Agilent) to ensure a DNA Integrity Number (DIN) >7 (value for NA12878 was 9.3). Concentration of DNA was assessed using the dsDNA HS assay on a Qubit fluorometer (Thermo Fisher).
Library preparation (SQK-LSK108 1D ligation genomic DNA).
1.5–2.5 μg human genomic DNA was sheared in a Covaris g-TUBE centrifuged at 5,000–6,000 r.p.m. in an Eppendorf 5424 (or equivalent) centrifuge for 2 × 1 min, inverting the tube between centrifugation steps.
DNA repair (NEBNext FFPE DNA Repair Mix, NEB M6630) was performed on purchased DNA but not on freshly extracted DNA. 8.5 μl nuclease-free water (NFW), 6.5 μl FFPE Repair Buffer and 2 μl FFPE DNA Repair Mix were added to the 46 μl sheared DNA. The mixture was incubated for 15 min at 20 °C, cleaned up using a 0.4× volume of AMPure XP beads (62 μl), incubated at room temperature with gentle mixing for 5 min, washed twice with 200 μl fresh 70% ethanol, pellet allowed to dry for 2 min, and DNA eluted in 46 μl NFW or EB (10 mM Tris pH 8.0). A 1 μl aliquot was quantified by fluorometry (Qubit) to ensure ≥1 μg DNA was retained.
End repair and dA-tailing (NEBNext Ultra II End-Repair/dA-tailing Module) was then performed by adding 7 μl Ultra II End-Prep buffer, 3 μl Ultra II End-Prep enzyme mix, and 5 μl NFW. The mixture was incubated at 20 °C for 10 min and 65 °C for 10 min. A 1× volume (60 μl) AMPure XP clean-up was performed and the DNA was eluted in 31 μl NFW. A 1-μl aliquot was quantified by fluorometry (Qubit) to ensure ≥700 ng DNA was retained.
Ligation was then performed by adding 20 μl Adaptor Mix (SQK-LSK108 Ligation Sequencing Kit 1D, Oxford Nanopore Technologies (ONT)) and 50 μl NEB Blunt/TA Master Mix (NEB, cat. no. M0367) to the 30 μl dA-tailed DNA, mixing gently and incubating at room temperature for 10 min.
The adaptor-ligated DNA was cleaned up by adding a 0.4 × volume (40 μl) of AMPure XP beads, incubating for 5 min at room temperature and resuspending the pellet twice in 140 μl ABB (SQK-LSK108). The purified-ligated DNA was resuspended by adding 25 μl ELB (SQK-LSK108) and resuspending the beads, incubating at room temperature for 10 min, pelleting the beads again, and transferring the supernatant (pre-sequencing mix or PSM) to a new tube. A 1-μl aliquot was quantified by fluorometry (Qubit) to ensure ≥500 ng DNA was retained.
Sambrook and Russell DNA extraction.
This protocol was modified from Chapter 6 protocol 1 of Sambrook and Russell51. 5 × 107 cells were spun at 4500g for 10 min to pellet. The cells were resuspended by pipette mixing in 100 μl PBS. 10 ml TLB was added (10 mM Tris-Cl pH 8.0, 25 mM EDTA pH 8.0, 0.5% (w/v) SDS, 20 μg/ml Qiagen RNase A), vortexed at full speed for 5 s and incubated at 37 °C for 1 h. 50 μl Proteinase K (Qiagen) was added and mixed by slow inversion ten times followed by 3 h at 50 °C with gentle mixing every 1 h. The lysate was phenol-purified using 10 ml buffer saturated phenol using phase-lock gel falcon tubes, followed by phenol:chloroform (1:1). The DNA was precipitated by the addition of 4 ml 5 M ammonium acetate and 30 ml ice-cold ethanol. DNA was recovered with a glass hook followed by washing twice in 70% ethanol. After spinning down at 10,000g, ethanol was removed followed by 10 min drying at 40 °C. 150 μl EB (Elution Buffer) was added to the DNA and left at 4 °C overnight to resuspend.
Library preparation (SQK-RAD002 genomic DNA).
To obtain ultra-long reads, the standard Rapid Adapters (RAD002) protocol (SQK-RAD002 Rapid Sequencing Kit, ONT) for genomic DNA was modified as follows. 16 μl of DNA from the Sambrook extraction at approximately 1 μg/μl, manipulated with a cut-off P20 pipette tip, was placed in a 0.2 ml PCR tube, with 1 μl removed to confirm quantification value. 5 μl FRM was added and mixed slowly ten times by gentle pipetting with a cut-off pipette tip moving only 12 μl. After mixing, the sample was incubated at 30 °C for 1 min followed by 75 °C for 1 min on a thermocycler. After this, 1 μl RAD and 1 μl Blunt/TA ligase was added with slow mixing by pipetting using a cut-off tip moving only 14 μl ten times. The library was then incubated at room temperature for 30 min to allow ligation of RAD. To load the library, 25.5 μl RBF (Running Buffer with Fuel mix) was mixed with 27.5 μl NFW, and this was added to the library. Using a P100 cut-off tip set to 75 μl, this library was mixed by pipetting slowly five times. This extremely viscous sample was loaded onto the “spot on” port and entered the flow cell by capillary action. The standard loading beads were omitted from this protocol owing to excessive clumping when mixed with the viscous library.
MinION sequencing.
MinION sequencing was performed as per manufacturer's guidelines using R9/R9.4 flow cells (FLO-MIN105/FLO-MIN106, ONT). MinION sequencing was controlled using Oxford Nanopore Technologies MinKNOW software. The specific versions of the software used varied from run to run but can be determined by inspection of fast5 files from the data set. Reads from all sites were copied off to a volume mounted on a CLIMB virtual server (http://www.climb.ac.uk) where metadata was extracted using poredb (https://github.com/nickloman/poredb) and base-calling performed using Metrichor (predominantly workflow ID 1200, although previous versions were used early on in the project) (http://www.metrichor.com). We note that base-calling in Metrichor has now been superseded by Albacore and is no longer available. Scrappie (https://github.com/nanoporetech/scrappie) was used for the chr20 comparisons using reads previously identified as being from this chromosome after mapping the Metrichor reads. Albacore 0.8.4 (available from the Oxford Nanopore Technologies user community) was used for the ultra-long read set, as this software became the recommended base-caller for nanopore reads in March 2017. Given the rapid development of upgrades to base-caller software we expect to periodically re-base-call these data and make the latest results available to the community through the Amazon Open Data site.
Modified MinION running scripts.
In a number of instances, MinION sequencing control was shifted to customized MinKNOW scripts. These scripts provided enhanced pore utilization/data yields during sequencing, and operated by monitoring and adjusting flow cell bias-voltage (–180 mV to –250 mV), and used an event-yield-dependent (70% of initial hour in each segment) initiation of active pore channel assignment via remuxing (reselection of ideal pores for sequencing from each group of four wells available around each channel on the flowcell). More detailed information on these scripts can be found on the Oxford Nanopore Technologies user community. In addition, a patch for all files required to modify MinION running scripts compatible with MinKNOW 1.3.23 only is available (Supplementary Code 1).
Live run monitoring.
To assist in choosing when to switch from a standard run script to a modified run protocol, a subset of runs was monitored with the assistance of the minControl tool, an alpha component of the minoTour suite of MinION run and analysis tools (https://github.com/minoTour/minoTour). minControl collects metrics about a run directly from the grouper software, which runs behind the standard ONT MinKNOW interface. minControl provides a historical log of yield measured in events from a flow cell enabling estimations of yield and the decay rate associated with loss of sequencing pores over time. MinKNOW yield is currently measured in events and is scaled by approximately 1.7 to estimate yield in bases.