2018年SCI论文--整合GEO数据挖掘完整复现 七 :DAVID在线工具进行KEGG富集分析
文章目录
-
- 论文地址
- DAVID官网
-
- 获得KEGG富集分析结果
- 气泡图
- cytoscape软件绘制代谢通路网络图
-
- 准备输入文件
-
- network data
- table data
- 输入network文件
- 输入table文件
- 设置风格、颜色
- 手动调整、保存图片
论文地址
DAVID官网
KEGG富集分析和GO富集分析方法一致,具体步骤见我上篇文章DAVID在线工具进行GO富集分析,这里主要展示可视化结果
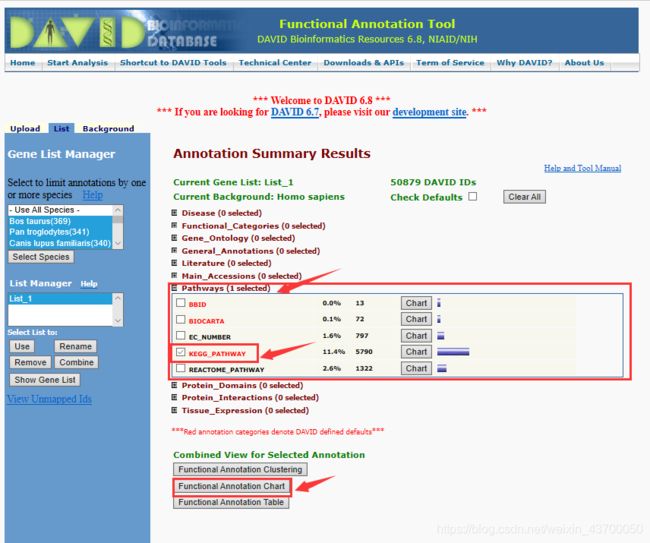
获得KEGG富集分析结果
1.输入文件为所有差异表达基因列表
2.选择GO富集分析结果时,我们点击“Pathways (1 selected)”下拉选项,选择“KEGG_PATHWAY”选项,最后保存为"kegg_pathway.txt"文件

气泡图
setwd("./3.DAVID_GO_KEGG/KEGG")
rt = read.table(file = 'kegg_pathway.txt',sep = '\t',header = T,quote = '')
keggSig = rt[rt$PValue < 0.05,]
library(tidyr)
keggSig = separate(keggSig, Term, sep = ":",
into = c("ID", "Term"))
library(ggplot2)
ggplot(keggSig,aes(x=Fold.Enrichment,y=Term)) +
geom_point(aes(size=Count,color=-1*log10(PValue)))+
scale_colour_gradient(low="green",high="red")+
labs(
color=expression(-log[10](P.value)),
size="Gene number",
x="Fold enrichment"
# y="Pathway name",
# title="Pathway enrichment")
)+
theme_bw()+
theme(
axis.text.y = element_text(size = rel(1.3)),
axis.title.x = element_text(size=rel(1.3)),
axis.title.y = element_blank()
)
ggsave('plot.pdf',width = 7,height = 4)

cytoscape软件绘制代谢通路网络图
软件下载地址,配置环境为java8,软件会自动下载安装java8,如果装了java9需要卸载后再安装,cytoscape教学视频
准备输入文件
network data
library(tidyr)
separate_keggSig = separate_rows(keggSig,Genes)
network_data = separate_keggSig[,c('ID','Genes')]
colnames(network_data)[2] = 'Name'
write.table(network_data,
file = 'network_data.txt',
sep = '\t',
quote = F,
row.names = F,
col.names = T)
table data
uniGene = unique(network_data$Name)
allSign = read.table(file = 'allSign.xls',sep = '\t',header = T,quote = '')
pathway_gene = allSign[match(uniGene,allSign$Name),]
pathway_gene$status = ifelse(pathway_gene$logFC > 0,'up','down')
df = data.frame(Name = keggSig[,2],status = rep('id',nrow(keggSig)))
table_data = rbind(df,pathway_gene[,c(1,3)])
write.table(table_data,
file = 'table_data.txt',
sep = '\t',
quote = F,
row.names = F,
col.names = T)
输入network文件

输入table文件
默认设置就好

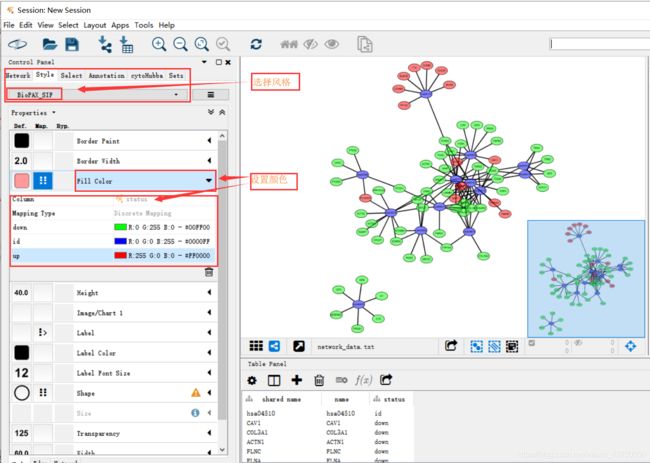
设置风格、颜色
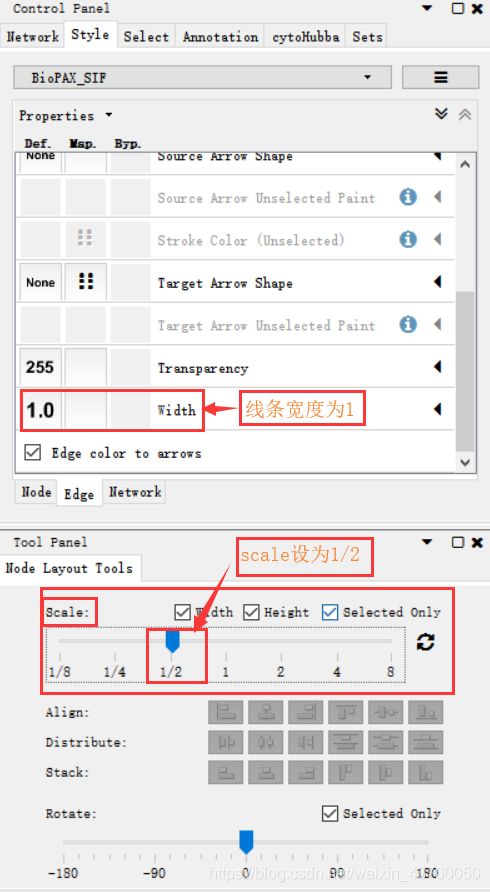
手动调整、保存图片
这个比较花时间,适合边听歌边画,还挺有意思的,哈哈
下面就是成品了,还是挺难看的,果然没有艺术细胞,一般图片保存为pdf格式,也可以保存为cys文件,方便再次在cytoscape中打开

本博客内容将同步更新到个人微信公众号:生信玩家。欢迎大家关注~~~