二代测序技术之illumina测序技术原理简介

现今的生信领域几乎就是和无数的序列打交道,而这些序列的来源就是如今风靡的高通量测序技术,现今的测序不论是测RNA、DNA、miRNA还是ChIP-Seq等等,都是基于NGS(二代测序,next-generation sequencing)的技术发展而来的,目前最为常用的就是illumina公司的测序技术,当然除了illumina公司外还是有其它二代测序技术存在的,ABI公司也有SOLiD测序技术。而之所以被称为二代测序,是因为比一代测序技术提升了非常多,具体体现在速度、准确性、效率等等方面。
由于序列对于生信行业的重要性,我认为了解基本的测序技术的原理是非常必要的,这也有利于生信技术人员了解到测序序列的特点和测序序列中可能出现的问题,了解到现今测序技术的局限性,甚至提出意见改进测序技术。因此本文会简要介绍当今最常用的illumina测序技术的测序原理。
文章目录
-
- 基因文库制备(sample prep)
- 生成序列簇(cluster generation)
- 测序(sequencing)
- 数据处理(data analysis)
- 叮
基因文库制备(sample prep)
制备测序基因文库是最初始的一步,这里我不过多解释文库,简要解释文库就是一个包含你所要测序的所有基因序列的集合体。测序过程中的基因文库的制备是将要测序的样本经过序列片段化(fragmentation)、再将这些片段化后得到的所有短序列的两端加上接头,从而将要测序的序列制备成为一个有很多拥有相同双端序列,但内部序列不同的序列片段集合体。具体步骤如下图。
图自:https://max.book118.com/html/2017/0924/134913788.shtm

解释一下图中的步骤:
- 首先将要测序的序列或者基因组等使用nebulisation(雾化)或者sonification(超声)再或者是酶学方法处理,即可得到片段化后的DNA序列,每个片段大概200-300bp。
- 片段化后的序列很大几率是拥有末端凸出的序列,并不是完整的配对状态,因此需要使用酶将其末端补平。
- 再使用酶在序列的平末端两端各加上一个A(腺嘌呤)尾。
- 通过上一步加上的A尾,illumina公司制作的接头(adapter)很容易加在序列的两端。
- 通过上面的步骤,制作步骤就结束了。但是需要提升测序序列的数量从而提高测序的准确性,也使后续的序列拼接步骤较为方便。因此最后还需要进行PCR操作扩增基因文库中序列的数量。由于每个序列的两端都有接头,因此设计的引物只需要和接头配对即可,十分方便。
有博客中写到文库制作是有两次序列片段化的过程的,这个具体我不是很清楚,但是实际上操作是一样的,只是会重复某些步骤而已。有兴趣的人可以去看这篇文章
这里要提一下加上的接头序列,一个序列片段加上接头后即变成如图的序列:
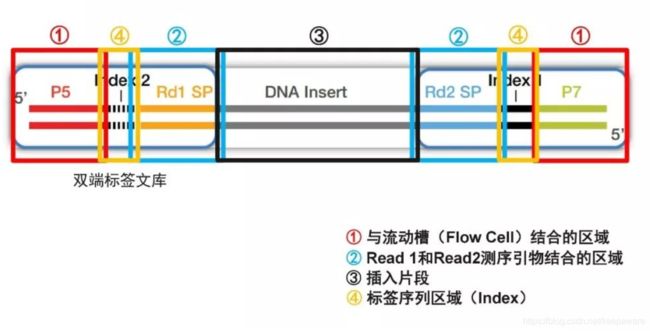
图自:https://www.cn-healthcare.com/articlewm/20210110/content-1179325.html
- 原始的序列在③处,而③之外的两端各为一个接头。
- ①处为后续会使用到的区域,暂时先不解释。
- ②处是测序时会使用到的片段,测序时发挥功能的引物是和②处配对的,配对后向中间部分(即③,要测序的序列)扩增序列,从而达到边合成、边测序(sequencing by synthesis)的效果。
- 这里图中提到的read1和read2是代表从正向和反向两次测序得到的序列结果。既然只是一条DNA序列,那为什么要测两次呢,这是因为有时候序列片段化不够彻底导致序列较长,而测序的序列若是较长则会导致测序的结果越来越不准,并且测序也可能达不到序列的全长,而从两端都进行测序就能够延长测序长度同时还能够使测序结果相对更为准确一些。
- ④处的序列是后续用于区别样品来源的一段引物配对序列。由于现今的测序技术已经达到很快速且很高效的地步,要是每次测序都只是测很短的序列,那么会导致资源浪费,因此每次测序可以尽量多测几组从而不浪费测序资源。这里的index序列就是后续区别所添加的样本来源的依据。后面测序步骤时会再介绍。
生成序列簇(cluster generation)
这一个步骤很多博客直接起名为桥式PCR,我觉得不够完整,桥式PCR只是是其中的一个重要的步骤,重要的是通过一系列包含了桥式PCR的步骤,从而生成了测序序列的序列簇。什么叫序列簇呢?就是相同的序列都聚集在一起从而形成的一簇序列。这个步骤是最最最核心的步骤,也是illumina公司的专利技术所在。
生成序列簇的意义是因为若是仅有一条序列进行测序,而测序时每条序列上又仅有一个荧光分子,而一个荧光分子所释放的荧光是很难被设备检测到的,而将每条需要测序的序列进行处理从而生成序列簇后就能在每次检测荧光时检测到一群相同的序列所释放的荧光,这样就不会有刚刚的问题了。大致了解序列簇后,接下来就要介绍序列簇的生成了。
序列簇的生成是在flowcell(流动槽)上形成的,一个flowcell的外观如图:

可以看到上面有八条白色的槽,这些槽也被成为阵列式流动槽,这些流动槽中使用illumina的技术从而使数十亿个纳米井按照固定位置分布在槽中,这样大数量的纳米井的存在使得数据产出增加、成本降低、运行时间也相对缩短了。接下来看一下纳米井是什么吧!
上面这张图就是flowcell放大非常多倍后的情况,可以看到上面密布着密密麻麻的孔,这些孔就是一个一个的纳米井,而每一个纳米井中的情况是:
上面这张图就是一个孔中的情况,每个孔中都密布着DNA探针,这些探针序列是能够和之前提到的接头序列互补配对的,所以每个孔中就只有两种DNA探针,这些探针序列是通过共价键结合到孔底部的(这些技术都是illumina的专利技术)。
了解了上面的知识后,就可以开始介绍如何制作序列簇啦。
首先,将之前制作好的DNA文库变性解旋,以使加入的序列都是单链状态,将这些单链序列加入到流动槽之中,而illumina独有的技术能够确保每个纳米井中仅进入一条序列。由于每个纳米井中只进入一条序列,因此每个纳米井中最终都只生成一个序列簇,也正是因此每一个纳米井中的所有序列发出的荧光都是一样的,因此对一个纳米井中的荧光检测的结果也就是一条序列测序的结果,这样也就完成对单独的一条序列进行测序的需求啦。
每条序列进入纳米井后,由于具有接头序列,因此其能够与纳米井中的一条探针序列配对,配对后加入dNTP和聚合酶就能够使序列延伸到文库中序列的长度(包含接头),如下图:

形成完整的双链后,再使序列变性解旋,而这时新合成的链由于其底部的探针序列是依靠共价键和底部相连的,因此其不会被洗脱掉。由于其不会被洗脱,其就能够依靠其伸出去的一端与附近的探针序列配对,如图:

就这样配对后,再加入dNTP和聚合酶,其又会被延伸,延伸后如图:

之后再变性解旋,就得到这样的两条链:

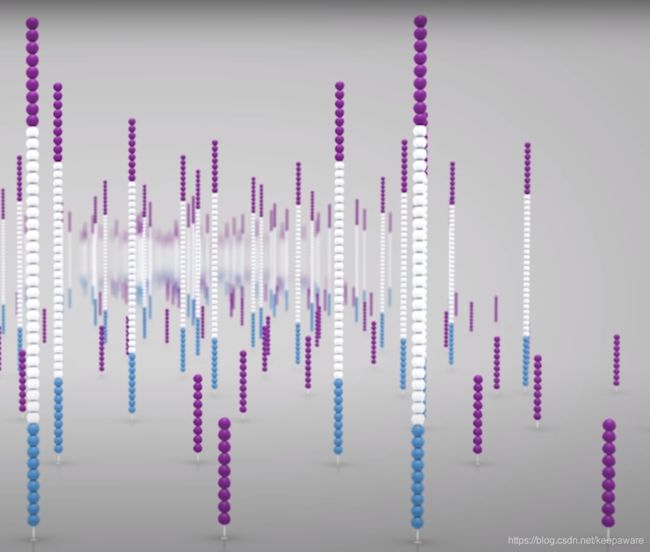
得到这样的两条链后,就反复地重复上面的桥式PCR的过程,从而使得每个纳米井中有非常多序列的拷贝数,才能达到测序的要求,如下图:

测序(sequencing)
达到上图所示的状态后,就差不多算完成了。但是要进行测序,就得先确定测序的方向,而刚刚提到了双端测序,也就是我们要对序列的两条单链序列进行分别测序。首先进行read1的测序,使用一种酶在能与read2引物配对的序列的某个位置上将read1切断,这样每个纳米井中就只剩下了一种方向的序列(不是5→3就是3→5),之后还会对伸出那端的核酸进行修饰防止额外延伸。这样就能够进行一端的测序啦。
如上图就可以开始测序啦,测序时先加入测序引物1,配对如图:

这样在体系中添加含有荧光标记的dNTP就开始测序了,添加有荧光标记的dNTP还额外包含一个叠氮基团,有叠氮基团的存在序列不能正常延伸,因此每添加上一个核酸后,序列的延伸会停止,这时候就能够在观测设备下根据纳米井发出的荧光颜色读取到正在合成的核酸,在观测后,使用特定酶水解调叠氮基团和荧光基团,这样下一个dNTP就能够正常进入到延伸序列中。就是不断的重复上面的步骤,最终就能够读取到序列的组成了。
贴一下示意图:
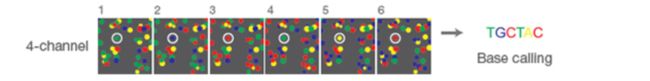
上面的图就是一个示意图,其中的一个固定的纳米井在不同的图中显示不同的颜色,就一一对应着序列信息。上图中使用的是四通道测序(即每种碱基是不同的荧光颜色,因此其需要收集四张照片,四张照片是因为每种荧光都需要分开去激发),现在illumina又有新的双通道测序技术了,双通道测序只依赖两种荧光颜色即可,有兴趣可以去了解一下。
上面第一部分介绍到了接头中包含了一端index序列,在测read1结束后就会测index序列了,测index序列时,首先需要将测序得到的双链水解掉。再使用read2引物和序列接头(这时配对的部分与read1部分不同,但是延伸的方向)配对,配对后延伸大概六到八个碱基即可,这六到八个碱基就能够用于确定样本的来源了。
测完index序列后就可以开始对read2的测序啦,要对read2测序很简单,就只需要再进行一次桥式PCR即可,这样得到的纳米井就是所有探针序列都被延伸过的结果,如图(同上面的一张图一样):
这时候再对能与read1引物配对的序列的某个位置的核酸进行切断处理就可以啦,就留下另外一条需要测序的链了。之后测序的步骤同之前测read1一样。
数据处理(data analysis)
在测序结束后,需要对得到的结果进行处理,这个方面我不是很清楚,不过可能大多会得到fastq文件。

fastq文件格式如上,
- 第一行是测序reads的ID以及其他信息。
- 第二行是序列
- 第三行以+开头,跟随着该read的名称。
- 第四行代表着每个碱基的测序质量。
这个部分不多讲,因为我不是很熟,以后有机会会补上的。
叮
参考:https://www.bilibili.com/video/BV13p411f7vx
参考:http://41j.com/blog/2012/04/nextgen-sequencing-primer/
参考:https://www.illumina.com.cn/science/technology/next-generation-sequencing/sequencing-technology/patterned-flow-cells.html?langsel=/cn/
参考:https://max.book118.com/html/2017/0924/134913788.shtm
参考:https://www.cn-healthcare.com/articlewm/20210110/content-1179325.html