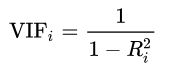- 系统学习Python——并发模型和异步编程:进程、线程和GIL
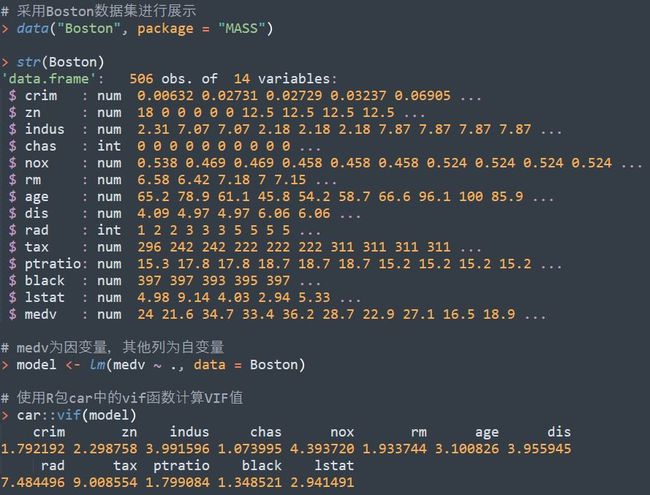
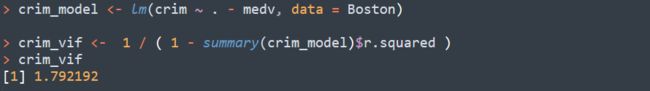
分类目录:《系统学习Python》总目录在文章《并发模型和异步编程:基础知识》我们简单介绍了Python中的进程、线程和协程。本文就着重介绍Python中的进程、线程和GIL的关系。Python解释器的每个实例都是一个进程。使用multiprocessing或concurrent.futures库可以启动额外的Python进程。Python的subprocess库用于启动运行外部程序(不管使用何种
- JavaScript 树形菜单总结
Auscy
microsoft
树形菜单是前端开发中常见的交互组件,用于展示具有层级关系的数据(如文件目录、分类列表、组织架构等)。以下从核心概念、实现方式、常见功能及优化方向等方面进行总结。一、核心概念层级结构:数据以父子嵌套形式存在,如{id:1,children:[{id:2}]}。节点:树形结构的基本单元,包含自身信息及子节点(若有)。展开/折叠:子节点的显示与隐藏切换,是树形菜单的核心交互。递归渲染:因数据层级不固定,
- 精通Canvas:15款时钟特效代码实现指南
烟幕缭绕
本文还有配套的精品资源,点击获取简介:HTML5的Canvas是一个用于绘制矢量图形的API,通过JavaScript实现动态效果。本项目集合了15种不同的时钟特效代码,帮助开发者通过学习绘制圆形、线条、时间更新、旋转、颜色样式设置及动画效果等概念,深化对Canvas的理解和应用。项目中的CSS文件负责时钟的样式设定,而JS文件则包含实现各种特效的逻辑,通过不同的函数或类处理时间更新和动画绘制,提
- 高效批量单词翻译工具的设计与应用
本文还有配套的精品资源,点击获取简介:在信息技术飞速发展的今天,批量单词翻译工具通过计算机的数据处理能力,大大提高了语言学习和文字处理的效率。用户通过简单输入单词列表到一个文本文件,并运行翻译程序,即可获得翻译结果并保存至指定文件。该工具集成了内置或外部翻译引擎,利用自然语言处理技术实现快速准确的翻译,并可能提供词性识别等附加功能。尽管机器翻译无法完全取代人工校对,但它为用户提供了一种高效的翻译解
- 嵌入式系统LCD显示模块编程实践
本文还有配套的精品资源,点击获取简介:本文档提供了一个具有800x480分辨率的3.5英寸液晶显示模块LW350AC9001的驱动程序代码,以及嵌入式系统中使用C/C++语言进行硬件编程的实践指南。该模块的2mm厚度使其适用于空间受限的便携式设备。内容包括驱动程序源代码、硬件控制接口使用方法,以及如何在嵌入式系统中进行图形处理、电源管理与性能优化。1.嵌入式系统原理1.1嵌入式系统概念嵌入式系统是
- 深入剖析OpenJDK 18 GA源码:Java平台最新发展
想法臃肿
本文还有配套的精品资源,点击获取简介:OpenJDK18GA作为Java开发的关键里程碑,提供了诸多新特性和改进。本文章深入探讨了OpenJDK18GA源码,揭示其内部机制,帮助开发者更好地理解和利用这个版本。文章还涵盖了PatternMatching、SealedClasses、Records、JEP395、JEP406和JEP407等特性,以及HotSpot虚拟机、编译器、垃圾收集器、内存模型
- FPGA小白到项目实战:Verilog+Vivado全流程通关指南(附光学类岗位技能映射)
阿牛的药铺
算法移植部署fpga开发verilog
FPGA小白到项目实战:Verilog+Vivado全流程通关指南(附光学类岗位技能映射)引言:为什么这个FPGA入门路线能帮你快速上岗?本文设计了一条**"Verilog语法→工具链操作→光学项目实战→岗位技能对标"的阶梯式学习路径。不同于泛泛而谈的FPGA教程,我们聚焦光学类产品开发**核心能力(时序接口设计、图像处理算法移植、高速接口应用),通过3个递进式项目(从LED闪烁到图像边缘检测),
- (Python基础篇)了解和使用分支结构
EternityArt
基础篇python
目录一、引言二、Python分支结构的类型与语法(一)if语句(单分支)(二)if-else语句(双分支)(三)if-elif-else语句(多分支)三、分支结构的应用场景(一)提示用户输入用户名,然后再提示输入密码,如果用户名是“admin”并且密码是“88888”则提示正确,否则,如果用户名不是admin还提示用户用户名不存在,(二)提示用户输入用户名,然后再提示输入密码,如果用户名是“adm
- vue keep-alive标签的运用
keep-alive,想必大家都不会很陌生,在一些选项卡中会使用到。其实,它的作用大概就是把组件的数据给缓存起来。比如果我有一个选项卡,标签一,标签二,标签三。现在,我需要实现,当我在标签一的表单中输入内容后,点击标签二,再回到标签一,表单的内容依然存在。如果按以往的做法,不使用keep-alive,那是不能实现的。然而,我们只需要在选项卡的内容最外层包一个keep-alive标签即可。但这儿有一
- 配音助手:自媒体神器,内置海量音色的语音,支持多主播配音
阿幸软件杂货间
媒体
软件介绍内置文字转语音,提供多个主播音色,男声、女声、小孩、方言。支持的场景也是比较多,比如:广告促销、有声读物、广播配音、影视配音、Ai配音等。这个软件是免费的,只不过需要通过手机号码登录就可以使用全部功能了。软件下载夸克下载
- centos7安装 mysql5.7(安装包)
heiPony
linuxmysqlmariadbcentosmysql
一.卸载centos7自带数据库查看系统自带的Mariadbrpm-qa|grepmariadbmariadb-libs-5.5.44-2.el7.centos.x86_64卸载rpm-e--nodepsmariadb-libs-5.5.44-2.el7.centos.x86_64删除etc目录下的my.cnfrm/etc/my.cnf二.检查mysql是否存在(有就卸载,删除相关文件)rpm-q
- Anaconda 详细下载与安装教程
Anaconda详细下载与安装教程1.简介Anaconda是一个用于科学计算的开源发行版,包含了Python和R的众多常用库。它还包括了conda包管理器,可以方便地安装、更新和管理各种软件包。2.下载Anaconda2.1访问官方网站首先,打开浏览器,访问Anaconda官方网站。2.2选择适合的版本在页面中,你会看到两个主要的下载选项:AnacondaIndividualEdition:适用于
- python中 @注解 及内置注解 的使用方法总结以及完整示例
慧一居士
Pythonpython
在Python中,装饰器(Decorator)使用@符号实现,是一种修改函数/类行为的语法糖。它本质上是一个高阶函数,接受目标函数作为参数并返回包装后的函数。Python也提供了多个内置装饰器,如@property、@staticmethod、@classmethod等。一、核心概念装饰器本质:@decorator等价于func=decorator(func)执行时机:在函数/类定义时立即执行装饰
- Maya自定义右键菜单样例教程
holy-pills
本文还有配套的精品资源,点击获取简介:本文详细指导如何在Maya中通过脚本节点自定义右键菜单,增强工作效率和个性化工作环境。自定义右键菜单允许用户根据个人习惯调整菜单项,使之更加便捷。文章介绍了创建脚本节点、编写菜单脚本、关联菜单到视图以及保存和加载自定义菜单的具体步骤。同时提供了实际操作样例,帮助用户更好地理解和应用这一技巧。1.Maya自定义右键菜单的重要性Maya,作为三维动画制作的行业标准
- ssrf漏洞复现
ξ流ぁ星ぷ132
安全
目录基础环境查看phpinfo发现线索探测端口+gopher协议基础环境这里发现一些基础协议呗过滤掉了。但是有个提示的info,于是先看看查看phpinfo发现线索发现这台主机的地址了,于是猜测这个网段应该还有其他主机,试了一下172.21.0.1:80172.21.0.3:80果然如下(0.1是陷阱就不浪费时间了,)探测端口+gopher协议然后对这个172.21.0.3这个主机探测端口发现63
- 零信任落地难题:安全性与用户体验如何两全?
粤海科技君
安全零信任终端安全网络安全iOA
在零信任架构的实施过程中,平衡安全性与用户体验是企业数字化转型的核心命题。这一挑战的本质在于:既要通过「永不信任,持续验证」的安全机制抵御新型攻击,又要避免过度验证导致的效率损耗。一、矛盾根源:安全与体验的天然张力零信任的“永不信任”原则,本质上要求对每一次访问都进行动态评估,但这与用户对“便捷、流畅”的诉求存在天然冲突。例如:频繁的身份验证(如每次登录都需短信验证码)会打断工作节奏,某制造企业统
- 从《哪吒 2》看个人IP的破局之道|创客匠人
《哪吒2》以破竹之势登顶中国影史票房榜,不到9天票房突破62亿,观众自发为其“冲百亿”的热情,揭示了一个朴素却深刻的商业逻辑:IP的真正生命力,不在于短暂曝光,而在于用户愿意用行动投票的长期信任。这种逻辑,同样适用于2025年个人IP的增长突围。流量失效的真相:用户体验断层终结增长如今的IP运营者常陷入一个误区:疯狂追逐流量,却留不住用户。短视频投流成本翻倍,内容越做越多粉丝却不涨,好不容易成交的
- 无面试无offer? 你需要AI 求职co-pilot的帮助!
大家好啊,我写的开源免费求职AIco-pilot工具发布了v3.0.0,欢迎大家参与、使用!https://github.com/weicanie/prisma-ai一、项目介绍开源免费的求职co-pilot,自动化简历准备至offer到手的整个流程。优化您的项目、定制您的简历、为您匹配工作,并帮助您做好面试准备。二、核心价值prisma-ai旨在解决求职者在准备简历和寻找工作时最头疼的3个问题:
- RocketMQ 之死信队列
firepation
RocketMQrocketmq
在分布式消息系统中,消息的可靠传递和处理至关重要。然而,由于各种原因(如消息处理失败、消费超时等),一些消息可能无法被正常消费。这些无法被消费的消息如果不加以处理,会影响系统的稳定性和数据一致性。为了解决这一问题,RocketMQ提供了死信队列(DeadLetterQueue,DLQ)机制。本文将深入探讨RocketMQ的死信队列,包括其实现原理、应用场景以及使用示例。什么是死信队列?死信队列是一
- 被动降噪的概念及编程实现
CodeByte
人工智能算法javascript编程
被动降噪是指通过编程技术和算法,对输入的数据进行处理,以减少或消除其中的噪声。噪声可以是各种形式的干扰,例如来自传感器、通信信号或其他外部源的干扰。在本文中,我们将探讨被动降噪的意义以及如何使用编程来实现这一目标。被动降噪的意义:噪声对数据的准确性和可靠性产生负面影响。在许多应用领域,例如图像处理、音频处理和信号处理中,噪声的存在可能导致数据质量下降,使得后续的分析和处理变得困难。因此,被动降噪技
- 等保测评中的物联网设备安全评估
亿林数据
物联网安全网络安全等保测评
随着物联网(IoT)技术的飞速发展,物联网设备已经广泛应用于智能家居、智慧城市、工业自动化等多个领域,极大地提升了社会生产力和生活便利性。然而,随着IoT设备数量的激增,其安全性问题也日益凸显,成为我们必须面对的重要课题。在这一背景下,等级保护(等保)测评中的物联网设备安全评估显得尤为重要,它为我们提供了一个有效的安全评估和管理机制。一、物联网设备安全评估的重要性物联网设备的核心理念是实现物物相连
- docker0网卡没有ip一步解决
ξ流ぁ星ぷ132
tcp/ip网络服务器
正常查看ip的时候一直显示没有ip这里先删除docker0网卡iplinkdeletedocker0然后重启服务systemctlrestartdocker再次查看显示有ip了并且查看配置文件也是正常的cat/etc/docker/daemon.json{"registry-mirrors":["https://docker.m.daocloud.io","https://docker.imgdb
- javascript高级程序设计第3版——第12章 DOM2与DOM3
weixin_30687587
javascript数据结构与算法ViewUI
12章——DOM2与DOM3为了增强D0M1,DOM级规范定义了一些模块。DOM2核心:为不同的DOM类型引入了一些与XML命名空间有关的方法,还定义了以编程方式创建Document实例的方法;DOM2级样式:针对操作元素的样式而开发;其特性总结:1.每个元素都有一个关联的style对象,可用来确定和修改行内样式;2.要确定某个元素的计算样式,可使用getComgetComputedStyle()
- JavaScript 基础09:Web APIs——日期对象、DOM节点
梦想当全栈
JavaScriptjavascript前端开发语言
JavaScript基础09:WebAPIs——日期对象、DOM节点进一步学习DOM相关知识,实现可交互的网页特效能够插入、删除和替换元素节点。能够依据元素节点关系查找节点。一、日期对象掌握Date日期对象的使用,动态获取当前计算机的时间。ECMAScript中内置了获取系统时间的对象Date,使用Date时与之前学习的内置对象console和Math不同,它需要借助new关键字才能使用。1.实例
- Excel控件Spire.XLS 更新至7.12.144 | 附下载
cocacola456
文档管理更新Excel控件Spire.XLS更新Spire.XLSSpire.XLS下载
Excel控件Spire.XLS更新至7.12.144,修复了转换PDF时字幕对齐的问题。Spire.XLS7.12.144更新修复修复了将Chart转换为Image时图表数据标签重复的问题。修复了CalculateAllValue方法抛出异常的问题。修复了将工作表转换为PDF时图表字幕对齐不正确的问题。
- Topview Avatar 2深度实测:AI数字人带货的新高度,还是又一个营销噱头?
神码小Z
AI工具人工智能
在AI数字人赛道越来越卷的今天,各家产品都在宣传自己的"独门秘技"。最近,TopviewAI推出的Avatar2引起了我的注意——号称突破了产品尺寸限制,实现了"万物皆可带"。作为一个经常需要制作营销视频的内容创作者,我决定亲自上手测试一番,看看这款工具是否真的像宣传的那样强大。TopviewAvatar2是什么?革命性升级还是渐进式改良?TopviewAvatar2是TopviewAI推出的第二
- C++设计秘籍:为什么所有参数都需类型转换时,非成员函数才是王道?
讳疾忌医丶
c++前端开发语言
当所有参数都需要类型转换时,为什么要选择非成员函数?在C++的世界里,有一个看似简单却蕴含深意的设计原则:当所有参数(包括被this指针所指的那个隐式参数)皆须进行类型转换时,请为此采用非成员函数实现。这个原则背后隐藏着C++类型系统的精妙设计,也揭示了成员函数与非成员函数在处理隐式类型转换时的本质差异。想象一下,你正在设计一个数学计算库,需要支持整数与有理数的混合运算。如果你天真地将所有操作都实
- C#中的设计模式:构建更加优雅的代码
Envyᥫᩣᩚ
c#开发语言
C#在面向对象编程(OOP)方面的强大支持,我们可以探讨“C#中的设计模式”。这不仅有助于理解如何更好地组织代码,还能提高代码的可维护性和可扩展性。引言设计模式是软件工程中经过实践验证的解决方案模板,它们提供了一种标准化的方法来解决常见的开发问题。对于使用C#进行开发的程序员来说,理解和应用这些模式可以帮助创建结构良好、易于维护和扩展的应用程序。本文将介绍几种常用的设计模式,并展示如何用C#实现它
- JAVA 高频八股文 Day03
Conqueror675
java开发语言
12.TCP和Http的区别是什么TCP是传输层协议,负责建立可靠的点对点连接,确保数据有序、完整地传输(如铁路轨道);HTTP是应用层协议,基于TCP构建,定义了Web服务交互的报文格式和规则(如货运订单)。TCP关注数据如何可靠送达,通过三次握手建立连接、流量控制等机制保证传输;HTTP关注传输内容的意义,提供请求/响应语义(GET/POST等)和无状态通信。补充:说一下什么是三次握手四次挥手
- 【证明】对极几何:本质矩阵内在性质
Powerful_QI
slam线性代数矩阵
--这是目录--1.本质矩阵内在性质表述2.预备知识2.1线性代数基础2.1.1奇异值与特征值的关系2.1.2矩阵加减单位阵后特征值的变化2.2引理:一个常用的矩阵变换3.证明1.本质矩阵内在性质表述 本质矩阵(EssentialMatrix)EEE是一个3阶方阵,满足E=t∧RE=t^{\land}RE=t∧R其中RRR为旋转矩阵,ttt为平移量,t∧t^{\land}t∧运算定义如下(参考了
- ios内付费
374016526
ios内付费
近年来写了很多IOS的程序,内付费也用到不少,使用IOS的内付费实现起来比较麻烦,这里我写了一个简单的内付费包,希望对大家有帮助。
具体使用如下:
这里的sender其实就是调用者,这里主要是为了回调使用。
[KuroStoreApi kuroStoreProductId:@"产品ID" storeSender:self storeFinishCallBa
- 20 款优秀的 Linux 终端仿真器
brotherlamp
linuxlinux视频linux资料linux自学linux教程
终端仿真器是一款用其它显示架构重现可视终端的计算机程序。换句话说就是终端仿真器能使哑终端看似像一台连接上了服务器的客户机。终端仿真器允许最终用户用文本用户界面和命令行来访问控制台和应用程序。(LCTT 译注:终端仿真器原意指对大型机-哑终端方式的模拟,不过在当今的 Linux 环境中,常指通过远程或本地方式连接的伪终端,俗称“终端”。)
你能从开源世界中找到大量的终端仿真器,它们
- Solr Deep Paging(solr 深分页)
eksliang
solr深分页solr分页性能问题
转载请出自出处:http://eksliang.iteye.com/blog/2148370
作者:eksliang(ickes) blg:http://eksliang.iteye.com/ 概述
长期以来,我们一直有一个深分页问题。如果直接跳到很靠后的页数,查询速度会比较慢。这是因为Solr的需要为查询从开始遍历所有数据。直到Solr的4.7这个问题一直没有一个很好的解决方案。直到solr
- 数据库面试题
18289753290
面试题 数据库
1.union ,union all
网络搜索出的最佳答案:
union和union all的区别是,union会自动压缩多个结果集合中的重复结果,而union all则将所有的结果全部显示出来,不管是不是重复。
Union:对两个结果集进行并集操作,不包括重复行,同时进行默认规则的排序;
Union All:对两个结果集进行并集操作,包括重复行,不进行排序;
2.索引有哪些分类?作用是
- Android TV屏幕适配
酷的飞上天空
android
先说下现在市面上TV分辨率的大概情况
两种分辨率为主
1.720标清,分辨率为1280x720.
屏幕尺寸以32寸为主,部分电视为42寸
2.1080p全高清,分辨率为1920x1080
屏幕尺寸以42寸为主,此分辨率电视屏幕从32寸到50寸都有
适配遇到问题,已1080p尺寸为例:
分辨率固定不变,屏幕尺寸变化较大。
如:效果图尺寸为1920x1080,如果使用d
- Timer定时器与ActionListener联合应用
永夜-极光
java
功能:在控制台每秒输出一次
代码:
package Main;
import javax.swing.Timer;
import java.awt.event.*;
public class T {
private static int count = 0;
public static void main(String[] args){
- Ubuntu14.04系统Tab键不能自动补全问题解决
随便小屋
Ubuntu 14.04
Unbuntu 14.4安装之后就在终端中使用Tab键不能自动补全,解决办法如下:
1、利用vi编辑器打开/etc/bash.bashrc文件(需要root权限)
sudo vi /etc/bash.bashrc
接下来会提示输入密码
2、找到文件中的下列代码
#enable bash completion in interactive shells
#if
- 学会人际关系三招 轻松走职场
aijuans
职场
要想成功,仅有专业能力是不够的,处理好与老板、同事及下属的人际关系也是门大学问。如何才能在职场如鱼得水、游刃有余呢?在此,教您简单实用的三个窍门。
第一,多汇报
最近,管理学又提出了一个新名词“追随力”。它告诉我们,做下属最关键的就是要多请示汇报,让上司随时了解你的工作进度,有了新想法也要及时建议。不知不觉,你就有了“追随力”,上司会越来越了解和信任你。
第二,勤沟通
团队的力
- 《O2O:移动互联网时代的商业革命》读书笔记
aoyouzi
读书笔记
移动互联网的未来:碎片化内容+碎片化渠道=各式精准、互动的新型社会化营销。
O2O:Online to OffLine 线上线下活动
O2O就是在移动互联网时代,生活消费领域通过线上和线下互动的一种新型商业模式。
手机二维码本质:O2O商务行为从线下现实世界到线上虚拟世界的入口。
线上虚拟世界创造的本意是打破信息鸿沟,让不同地域、不同需求的人
- js实现图片随鼠标滚动的效果
百合不是茶
JavaScript滚动属性的获取图片滚动属性获取页面加载
1,获取样式属性值
top 与顶部的距离
left 与左边的距离
right 与右边的距离
bottom 与下边的距离
zIndex 层叠层次
例子:获取左边的宽度,当css写在body标签中时
<div id="adver" style="position:absolute;top:50px;left:1000p
- ajax同步异步参数async
bijian1013
jqueryAjaxasync
开发项目开发过程中,需要将ajax的返回值赋到全局变量中,然后在该页面其他地方引用,因为ajax异步的原因一直无法成功,需将async:false,使其变成同步的。
格式:
$.ajax({ type: 'POST', ur
- Webx3框架(1)
Bill_chen
eclipsespringmaven框架ibatis
Webx是淘宝开发的一套Web开发框架,Webx3是其第三个升级版本;采用Eclipse的开发环境,现在支持java开发;
采用turbine原型的MVC框架,扩展了Spring容器,利用Maven进行项目的构建管理,灵活的ibatis持久层支持,总的来说,还是一套很不错的Web框架。
Webx3遵循turbine风格,velocity的模板被分为layout/screen/control三部
- 【MongoDB学习笔记五】MongoDB概述
bit1129
mongodb
MongoDB是面向文档的NoSQL数据库,尽量业界还对MongoDB存在一些质疑的声音,比如性能尤其是查询性能、数据一致性的支持没有想象的那么好,但是MongoDB用户群确实已经够多。MongoDB的亮点不在于它的性能,而是它处理非结构化数据的能力以及内置对分布式的支持(复制、分片达到的高可用、高可伸缩),同时它提供的近似于SQL的查询能力,也是在做NoSQL技术选型时,考虑的一个重要因素。Mo
- spring/hibernate/struts2常见异常总结
白糖_
Hibernate
Spring
①ClassNotFoundException: org.aspectj.weaver.reflect.ReflectionWorld$ReflectionWorldException
缺少aspectjweaver.jar,该jar包常用于spring aop中
②java.lang.ClassNotFoundException: org.sprin
- jquery easyui表单重置(reset)扩展思路
bozch
formjquery easyuireset
在jquery easyui表单中 尚未提供表单重置的功能,这就需要自己对其进行扩展。
扩展的时候要考虑的控件有:
combo,combobox,combogrid,combotree,datebox,datetimebox
需要对其添加reset方法,reset方法就是把初始化的值赋值给当前的组件,这就需要在组件的初始化时将值保存下来。
在所有的reset方法添加完毕之后,就需要对fo
- 编程之美-烙饼排序
bylijinnan
编程之美
package beautyOfCoding;
import java.util.Arrays;
/*
*《编程之美》的思路是:搜索+剪枝。有点像是写下棋程序:当前情况下,把所有可能的下一步都做一遍;在这每一遍操作里面,计算出如果按这一步走的话,能不能赢(得出最优结果)。
*《编程之美》上代码有很多错误,且每个变量的含义令人费解。因此我按我的理解写了以下代码:
*/
- Struts1.X 源码分析之ActionForm赋值原理
chenbowen00
struts
struts1在处理请求参数之前,首先会根据配置文件action节点的name属性创建对应的ActionForm。如果配置了name属性,却找不到对应的ActionForm类也不会报错,只是不会处理本次请求的请求参数。
如果找到了对应的ActionForm类,则先判断是否已经存在ActionForm的实例,如果不存在则创建实例,并将其存放在对应的作用域中。作用域由配置文件action节点的s
- [空天防御与经济]在获得充足的外部资源之前,太空投资需有限度
comsci
资源
这里有一个常识性的问题:
地球的资源,人类的资金是有限的,而太空是无限的.....
就算全人类联合起来,要在太空中修建大型空间站,也不一定能够成功,因为资源和资金,技术有客观的限制....
&
- ORACLE临时表—ON COMMIT PRESERVE ROWS
daizj
oracle临时表
ORACLE临时表 转
临时表:像普通表一样,有结构,但是对数据的管理上不一样,临时表存储事务或会话的中间结果集,临时表中保存的数据只对当前
会话可见,所有会话都看不到其他会话的数据,即使其他会话提交了,也看不到。临时表不存在并发行为,因为他们对于当前会话都是独立的。
创建临时表时,ORACLE只创建了表的结构(在数据字典中定义),并没有初始化内存空间,当某一会话使用临时表时,ORALCE会
- 基于Nginx XSendfile+SpringMVC进行文件下载
denger
应用服务器Webnginx网络应用lighttpd
在平常我们实现文件下载通常是通过普通 read-write方式,如下代码所示。
@RequestMapping("/courseware/{id}")
public void download(@PathVariable("id") String courseID, HttpServletResp
- scanf接受char类型的字符
dcj3sjt126com
c
/*
2013年3月11日22:35:54
目的:学习char只接受一个字符
*/
# include <stdio.h>
int main(void)
{
int i;
char ch;
scanf("%d", &i);
printf("i = %d\n", i);
scanf("%
- 学编程的价值
dcj3sjt126com
编程
发一个人会编程, 想想以后可以教儿女, 是多么美好的事啊, 不管儿女将来从事什么样的职业, 教一教, 对他思维的开拓大有帮助
像这位朋友学习:
http://blog.sina.com.cn/s/articlelist_2584320772_0_1.html
VirtualGS教程 (By @林泰前): 几十年的老程序员,资深的
- 二维数组(矩阵)对角线输出
飞天奔月
二维数组
今天在BBS里面看到这样的面试题目,
1,二维数组(N*N),沿对角线方向,从右上角打印到左下角如N=4: 4*4二维数组
{ 1 2 3 4 }
{ 5 6 7 8 }
{ 9 10 11 12 }
{13 14 15 16 }
打印顺序
4
3 8
2 7 12
1 6 11 16
5 10 15
9 14
13
要
- Ehcache(08)——可阻塞的Cache——BlockingCache
234390216
并发ehcacheBlockingCache阻塞
可阻塞的Cache—BlockingCache
在上一节我们提到了显示使用Ehcache锁的问题,其实我们还可以隐式的来使用Ehcache的锁,那就是通过BlockingCache。BlockingCache是Ehcache的一个封装类,可以让我们对Ehcache进行并发操作。其内部的锁机制是使用的net.
- mysqldiff对数据库间进行差异比较
jackyrong
mysqld
mysqldiff该工具是官方mysql-utilities工具集的一个脚本,可以用来对比不同数据库之间的表结构,或者同个数据库间的表结构
如果在windows下,直接下载mysql-utilities安装就可以了,然后运行后,会跑到命令行下:
1) 基本用法
mysqldiff --server1=admin:12345
- spring data jpa 方法中可用的关键字
lawrence.li
javaspring
spring data jpa 支持以方法名进行查询/删除/统计。
查询的关键字为find
删除的关键字为delete/remove (>=1.7.x)
统计的关键字为count (>=1.7.x)
修改需要使用@Modifying注解
@Modifying
@Query("update User u set u.firstna
- Spring的ModelAndView类
nicegege
spring
项目中controller的方法跳转的到ModelAndView类,一直很好奇spring怎么实现的?
/*
* Copyright 2002-2010 the original author or authors.
*
* Licensed under the Apache License, Version 2.0 (the "License");
* yo
- 搭建 CentOS 6 服务器(13) - rsync、Amanda
rensanning
centos
(一)rsync
Server端
# yum install rsync
# vi /etc/xinetd.d/rsync
service rsync
{
disable = no
flags = IPv6
socket_type = stream
wait
- Learn Nodejs 02
toknowme
nodejs
(1)npm是什么
npm is the package manager for node
官方网站:https://www.npmjs.com/
npm上有很多优秀的nodejs包,来解决常见的一些问题,比如用node-mysql,就可以方便通过nodejs链接到mysql,进行数据库的操作
在开发过程往往会需要用到其他的包,使用npm就可以下载这些包来供程序调用
&nb
- Spring MVC 拦截器
xp9802
spring mvc
Controller层的拦截器继承于HandlerInterceptorAdapter
HandlerInterceptorAdapter.java 1 public abstract class HandlerInterceptorAdapter implements HandlerIntercep