RNAseq分析全过程
本次实操数据来自下面的文章,是量化环境葡萄糖对转录组的影响,比较低糖和高糖环境下胰岛转录组的变化,胰岛来源小鼠。
SRR1374921 LoGlu_Rep1
SRR1374922 LoGlu_Rep2
SRR1374923 HiGlu_Rep1
SRR1374924 HiGlu_Rep2
该数据已发表文章:
The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression
DOI: 10.1186/1471-2164-15-62
详细分析过程
- 1 安装conda及RNA-seq所需要的工具
-
- 1.1 conda安装
- 1.2 SRA-toolkit
- 1.3 fastqc
- 1.4 fastp
- 1.5 hisat2
- 1.6 samtools
- 1.7 htseq
- 2 下载原始数据 参考基因组及其注释
-
- 2.1 原始数据
- 2.2 参考基因组及其注释
- 3 sra到fastq格式转换并进行质量控制
-
- 3.1 格式转换
- 3.2 质量控制
- 4 序列比对 Hisat2
- 5 reads计数 合并矩阵并进行注释
-
- 5.1 reads计数
- 5.2 合并矩阵并进行注释
- 6 DEseq2筛选差异表达基因并用bioMart注释
-
- 6.1 DEseq2筛选差异表达基因
- 6.2 用bioMart对差异表达基因进行注释
- 7 数据可视化 绘图
-
- 7.1 MA图
- 7.2 Plot counts
- 7.3 PCA
- 7.4 热图
- 7.5 火山图
- 8 富集分析 功能注释
- 9 KEGG通路可视化
1 安装conda及RNA-seq所需要的工具
1.1 conda安装
安装
#下载压缩包
wget https://repo.anaconda.com/archive/Anaconda3-2021.05-Linux-x86_64.sh
#安装
bash Anaconda3-2021.05-Linux-x86_64.sh
#配置环境变量
vim ~/.bashrc
export PATH="/root/weirdo/anaconda3/bin:$PATH"
#激活环境变量
source /etc/bash.bashrc
#测试是否安装成功 如果成功安装会出现conda的版本
conda --version
#如果第一次安装没有成功 第二次安装的时候需要在ENTER那一步修改安装路径 否则会因为安装路径被占用而无法安装成功
一些相关命令
查看变量 echo $PATH
用户配置文件 ~ /.bshrc
启动环境:source activate
添加镜像源:onda config -add conda config --show
查看已有环境:conda env -info
搜索:conda search
创建新环境:conda create -n env_name -prefix python=2 bwa
删除环境:conda remove -n env_name -all
配置python环境
#查看当前python版本号
python --version
#创建一个名为python27的环境 指定Python版本是2.7(不用管是2.7.x conda会为我们自动寻找2.7.x中的最新版本)
conda create --name python27 python=2.7 -y
#激活python环境 两种方式
conda activate python2
source activate pythion2
#查看当前python版本号
python --version
#列举当前所有环境
conda info -e
所需要的软件可以到生信技能树RNA-seq基础传送门里查看,(需要复制链接到浏览器中查看): http://www.biotrainee.com/thread-1750-1-1.html
1.2 SRA-toolkit
官网链接: https://ccb.jhu.edu/software/hisat2/index.shtml,有详细的使用说明。
1 预编译安装
#进入shares目录 这里是因为我的shares目录里已经下载好了 就直接切换进入解压缩
ll ../shares
#创建Biosofts目录
mkdir ~/Biosofts/
#解压文件 -C指定解压缩的目录文件
tar zxvf /disk1/shares/sratoolkit.2.11.1-ubuntu64.tar.gz -C ~/Biosofts/
#测试安装是否成功
~/Biosofts/sratoolkit.2.11.1-ubuntu64/bin/fastq-dump -h
#添加环境变量
echo 'export PATH=~/Biosofts/sratoolkit.2.11.1-ubuntu64/bin:$PATH' >>~/.bashrc
#使环境变量生效
source ~/.bashrc
#再次测试安装情况 两个都可以 如果成功会出现软件使用说明
fastq-dump
prefetch -h
#如果服务器里没有下载 则用wget下载 选择和电脑匹配的版本
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.9.2/sratoolkit.2.9.2-centos_linux64.tar.gz
2 root用户apt-get安装
sudo apt install sra-toolkit
prefetch -h
1.3 fastqc
FastQC是一款基于Java的软件,它可以快速地对测序数据进行质量评估,其官网为:链接:Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data
1 安装java环境
#创建文件目录
sudo mkdir /usr/java
mkdir /usr/java
#下载jdk压缩包
wget https://repo.huaweicloud.com/java/jdk/10.0.1+10/jdk-10.0.1_linux-x64_bin.tar.gz
#给相关目录创建软连接
sudo ln -s jdk1.8.0_172 latest
sudo ln -s /usr/java/latest default
#进入文件修改环境变量
sudo vi /etc/profile
#添加环境变量
export JAVA_HOME=/usr/java/latest
export PATH=JAVA_HOME/bin:JAVA_HOME/jre/bin:$PATH
export CLASSPATH=.:$JAVA_HOME/lib/dt.jar:JAVA_HOME/lib/tools.jar
#使修改的环境变量生效
source /etc/profile
#测试是否安装成功
java -version
2 安装fastqc
#切换到指定目录
cd ~/Biosofts
#下载fastq压缩包
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
#创建下一级目录
mkdir ~/Biosofts/fastqc
#解压文件到指定目录
unzip /disk1/shares/fastqc_v0.11.7.zip -d ~/Biosofts/
#给目录加上执行权限
chmod +x ~/Biosofts/FastQC/fastqc
#fastqc默认安装
~/Biosofts/FastQC/fastqc -h
#加入环境变量 就不用再输入路径了
echo 'export PATH=~/Biosofts/FastQC:$PATH'>>~/.bashrc
#使环境变量生效
source ~/.bashrc
#测试是否安装成功 如果成功会出现使用说明
fastqc -h
1.4 fastp
fastp是用于处理fastq文件,基于C++,支持多线程,包含fastQC和Trimmomatic的一些功能,运行速度比Trimmomatic快,并且可以同时过滤和质控,比较方便。
fastp功能
- 去接头
- 碱基矫正
- 滑动窗口质量值剪切
- 切ployG/ployX尾巴
- 处理分子标签(UMI)
- 分割输出结果
- duplicate率的评估
- 过表达序列分析
- 质控结果报告
fastp常用参数详细说明可参考:https://blog.csdn.net/sinat_32872729/article/details/94440265
conda安装
conda install fastp
1.5 hisat2
主要用于序列比对,一般用在RNA-seq里,使用方法: https://blog.csdn.net/narutodzx/article/details/126471422
1 源代码安装
#后台运行 就算界面卡住也能继续运行
screen
#拷贝文件 没有的话用wget下载
cp /disk1/shares/hisat2-2.2.0-source.zip ./
#解压文件
unzip hisat2-2.2.0-source.zip
#切换到hisat2-2.2.0目录
cd hisat2-2.2.0
#运行
make
2 预编译安装
#下载压缩包
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-Linux_x86_64.zip
#解压压缩文件
unzip hisat2-2.1.0-Linux_x86_64.zip
#进入文件夹
cd hisat2-2.2.0/
#运行hisat2命令
./hisat2
#设置环境变量 使其生效
echo 'export PATH=~/hisat2-2.2.0:$PATH' >> ~/.bashrc
source ~/.bashrc
#运行
hisat2
1.6 samtools
官方手册: http://www.htslib.org/doc/samtools.html
使用链接: http://quinlanlab.org/tutorials/samtools/samtools.html
sudo apt install samtools
samtools
samtools view
1.7 htseq
# 安装htseq
conda install htseq
htseq-count --v
2 下载原始数据 参考基因组及其注释
2.1 原始数据
原始数据一般是sra文件,后面再转换为fastq文件或sam文件,一般有三种下载方式:
- ascp下载(推荐数据较大时使用)
- sra-toolkit中prefetch下载
- wget下载
理论上下载方式推荐ascp>prefetch>wget,但因为目前NCBI网站不再提供ftp地址,需要到EBI-ENA数据库去寻找ftp地址,所以一般数据不是特别特别大的时候选择prefetch下载,wget下载可能会下载不全且花费时间较多。
SRA数据库相关知识可以看这个: https://blog.csdn.net/qq_22253901/article/details/120069582
ascp下载
1.在EBI-ENA数据库找到对应ftp地址
在搜索框输入检索号SRR*,点击Show Column Selection,选择run-accession,fastq-aspera,sra-aspera这三列,然后点击Down report的TSV格式,本地查看下载得到的filereport-read-run-SRR*-tsv.txt文件,会看到ftp地址,这个地址是ascp下载必须地址。

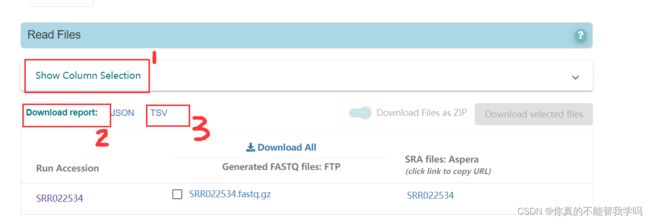
2.Linux里下载
这里的数据不是本次分析的数据,是为了举例说明ascp的使用,如采用此种办法,需要去寻找ftp地址,然后替换掉命令里的地址就可以。
mkdir SRX532741_sra/
screen
ascp -v -QT -l 400m -P33001 -k1 -i /root/.aspera/connect/etc/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list ENA_SRX5327410_sra_aspera.txt /data/SRX5327410_sra/
#这里的root/.aspera/connect/etc/asperaweb_id_dsa.openssh是自己该文件的路径
#/data/SRX5327410_sra/ 下载文件存储路径
#ENA_SRX5327410_sra_aspera.txt 若该文件在其他位置 必须是全路径
#-k1 断点续传
#-T 取消加密 否则有时候数据下载不了
#-l 设置最大传输速度 200~500m
#-Q 用于自适应流量控制 磁盘限制所需
#-i 提供私钥文件的地址 免密从SRA和ENA下载 此选项每次命令都需要加入 一般为~/.aspera/connect/etc/asperaweb_id_dsa.openssh(即-i参数后直接加此文件)
#-P 用于SSH身份验证的TCP端口 一般是33001
#--user 指定用户名 era-fasp是aspera在ENA下的用户名
#--host 跟的是固定下载地址asp.sra.ebi.ac.uk
prefetch下载
这里下载的数据是我分析所用的数据,一共有四个。
#下载单个文件
prefetch SRR1374921
#批量下载
mkdir data
cd data
touch data_download.sh
vim data_download.sh
#!/bin/bash
for i in 1 2 3 4
do
prefetch SRR137492${i}
done
#Esc : wq! 保存并退出编辑
chmod 777 data_download.sh
./data_download.sh
#fastq-dump下载 可直接得到fastq文件
fastq-dump --split-files SRR1374921.sra
fastq-dump --split-files SRR1374921
fastq-dump --gzip --split-3 SRR1374921
#--split-spot: 将双端测序分为两份 但是都放在同一个文件中
#--split-files: 将双端测序分为两份 放在不同的文件 但是对于一方有而一方没有的reads直接丢弃
#--split-3: 将双端测序分为两份 放在不同的文件 但是对于一方有而一方没有的reads会单独放在一个文件夹里
#--gzip: 输出文件为gzip压缩格式
wget下载
这个就比较简单直接在NCBI官网的搜索框输入SRR1374921,可以看到该菌的目前研究以及进展,在最下面Runs点击SRR1374921,在跳转界面点击Data access,会看到一个网址,复制网址,然后wget跟网址就可以,不过这种下载速度比较慢,所以最好放在screen窗口运行。
2.2 参考基因组及其注释
参考基因组下载
常用的三大网站:
- NCBI
- UCSC
- Ensemble
#UCSC官网下载小鼠参考基因组文件 mm39.fa 最后生成fai文件 sam转bam时用
#gencode官网下载小鼠基因组注释文件 gtf文件
#下载索引
cd ../
mkdir genome
cd genome
wget https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz #参考基因组文件
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M31/gencode.vM31.annotation.gtf.gz #基因组注释文件
wget https://cloud.biohpc.swmed.edu/index.php/s/grcm38_tran #索引文件
gunzip mm39.fa.gz
gunzip gencode.vM31.annotation.gtf.gz
tar -zxvf grcm38_tran.tar.gz
参考基因组文件:测序得到的是几百bp的短read, 相当于把拼图打散了给你。如果没有参考基因组,从头(de novo)组装等于是重走人类基因组计划的老路,也就是打散了拼图,却不告诉你原来是什么样子,那么任务将会及其艰巨,所以需要用到参考基因组。
基因组注释文件:相当于基因的说明书,告诉哪些是编码蛋白的基因,哪些是非编码基因,外显子,内含子,UTR位置等等,上面的三个网站都有注释文件,常用的是gencode数据库下载,在数据框选择小鼠复制对应版本的网址然后用wget下载。
参考文章:
https://www.jianshu.com/p/3e545b9a3c68
https://www.jianshu.com/p/849f8ada0ab7
3 sra到fastq格式转换并进行质量控制
3.1 格式转换
利用samtools中的fastq-dump将sra转换为fastq,fastq-dump中间没有空格,具体用法参考官网: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc&f=fastq-dump
cd data #切换到数据存放的目录
touch fastq_dump.sh
vim fastq_dump.sh
#!/bin/bash
for i in 1 2 3 4
do
echo SRR137492${i} #打印输出文件名
fastq-dump --gzip SRR137492${i}/SRR137492${i}.sra
#可根据数据需要选择--spilt-files/--spilt-3/--spilt-spot
done
#Esc : wq! 保存并退出
chmod 777 fastq_dump.sh
screen
./fastq_dump.sh
3.2 质量控制
我选择的是fastp质控,也可以选择fastqc质控,再用multiQC批量查看质控结果,fastqc的操作可以看着一篇: https://www.jianshu.com/p/facb4a1e5927
touch fastp.sh
vim fastp.sh
#!/bin/bash
for i in 1 2 3 4
do
fastp -i SRR137492${i}.fastq.gz -o SRR137492${i}.fq.gz
done
#Esc : wq! 保存并退出
chmod 777 fastp.sh
screen
./fastp.sh
#结果只看到一个fastp.html文件,可能前面的几个被覆盖了,可以设置参数解决
# -h --html 设置输出 html 格式的质控结果文件名 不设置则默认 html 文件名为 fastp.html
# -j --json 设置输出 html 格式的质控结果文件名 不设置则默认 json 文件名为 fastp.json
这里直接看最后一个质控结果,能够看到过滤前后的一些数据,也可以直观地看出很多指标,如过滤前后的 reads 数、bases 数、碱基质量、插入片段长度、碱基组成、GC 含量、接头类别等,具体意义可参考链接: https://blog.csdn.net/twocanis/article/details/109681242

4 序列比对 Hisat2
序列比对有很多软件,bwa,bowtie2,hisat2等,这里选择了hisat2,可以在这几篇文章找寻答案:
https://www.jianshu.com/p/681e02e7f9af
https://zhuanlan.zhihu.com/p/26506787
https://www.jianshu.com/p/849f8ada0ab7
#在data目录下
samtools faidx mm39.fa
touch hisat2_samtools.sh
vim hisat2_samtools.sh
#!/bin/bash
for i in 1 2 3 4;do
{
hisat2 -t -x /disk1/202031107010114/RNAseq/genome_index/mm10/genome -U /disk1/202031107010114/RNAseq/data/SRR137492${i}.fq.gz -S SRR137492${i}.sam;
samtools view -bhS -t /disk1/202031107010114/RNAseq/genome/mm39.fa.fai -o SRR137492${i}.bam SRR137492${i}.sam;
samtools sort SRR137492${i}.bam -o SRR137492${i}.sorted.bam;
samtools index SRR137492${i}.sorted.bam;}
done
#Esc : wq! 保存并退出
chmod 777 hisat2_samtools.sh
hisat2使用方法
Usage:
hisat2 [options]* -x {-1 -2 | -U | --sra-acc } [-S ]
Index filename prefix (minus trailing .X.ht2).
Files with #1 mates, paired with files in .
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
Files with #2 mates, paired with files in .
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
Comma-separated list of SRA accession numbers, e.g. --sra-acc SRR353653,SRR353654.
File for SAM output (default: stdout)
, , can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
-x :参考基因组索引文件的前缀
-1 :双端测序结果的第一个文件 若有多组数据 使用逗号将文件分隔 Reads的长度可以不一致
-2 :双端测序结果的第二个文件 若有多组数据 使用逗号将文件分隔 并且文件顺序要和-1参数对应 Reads的长度可以不一致
-U :单端数据文件 若有多组数据 使用逗号将文件分隔 可以和-1 -2参数同时使用 Reads的长度可以不一致
–sra-acc :输入SRA登录号 比如SRR353653 多组数据之间使用逗号分隔 HISAT将自动下载并识别数据类型 进行比对
-S :指定输出的SAM文件
samtools用法
官方说明和使用手册见前面安装部分的链接,这里说明samtools功能和参数意义。
view: BAM-SAM/SAM-BAM 转换和提取部分比对
sort: 比对排序
merge: 聚合多个排序比对
index: 索引排序比对
faidx: 建立FASTA索引 提取部分序列
tview: 文本格式查看序列
pileup: 产生基于位置的结果和 consensus/indel calling
-b:output BAM默认输出是SAM文件格式文件 该参数设置输出BAM格式
-h:默认输出文件不带header 设置后但header信息
-S:输入文件是SAM 加上避免报错
-t:使用list文件作为输入
5 reads计数 合并矩阵并进行注释
5.1 reads计数
# 首先将bam文件按reads名称进行排序(前期是按照默认的染色体位置进行排序的,所以要重新进行排序)
for i in 1 2 3 4;do samtools sort -n SRR137492${i}.bam -o SRR137492${i}.nsorted.bam;done
# htseq-count计数 用的是前面在genome里下载的注释文件 得到count文件
for i in 1 2 3 4;do htseq-count -r name -f bam /disk1/202031107010114/RNAseq/data/SRR137492${i}.nsorted.bam /disk1/202031107010114/RNAseq/genome_index/gencode.vM31.annotation.gtf > SRR137492${i}.count;done
# 计数
wc -l *.count
# 查看前几行和后几行
head -n 4 *.count
tail -n 4 *.count
5.2 合并矩阵并进行注释
做到这里前面已经完成了大部分啦,最煎熬地等待命令运行的过程也过去了,后面的操作基本都是在R中完成了,将上一步得到的四个count文件下载到本地,最好是直接放在RStudio的工作路径下,这样就不用专门再加绝对路径。
合并矩阵
#合并表达矩阵并进行注释(miomart)
#不需要变成因子
options(stringsAsFactors=FALSE)
#导入数据设置分隔符行名 设置分割符是\t 行名是gene.id和实验对应名称
LoGlu_Rep1<-read.table("D:/Users/hemiao/Desktop/shiyan/SRR1374921.count",sep="\t",col.names=c("gene_id","LoGlu_Rep1"))
head(control1)
LoGlu_Rep2<-read.table("D:/Users/hemiao/Desktop/shiyan/SRR1374922.count",sep="\t",col.names=c("gene_id","LoGlu_Rep2"))
HiGlu_Rep1<-read.table("D:/Users/hemiao/Desktop/shiyan/SRR1374923.count",sep="\t",col.names=c("gene_id","HiGlu_Rep1"))
HiGlu_Rep2<-read.table("D:/Users/hemiao/Desktop/shiyan/SRR1374924.count",sep="\t",col.names=c("gene_id","HiGlu_Rep2"))
#数据整合 merge函数 以gene_id将四组数据合并到一起
raw_count<-merge(merge(LoGlu_Rep1,LoGlu_Rep2,by="gene_id"),merge(HiGlu_Rep1,HiGlu_Rep2,by="gene_id"))
#删除前五行无关的 删除之前先查看一下是哪几行
raw_count_filt<-raw_count[-1:-5,]
#EBI数据库无法识别到小数点后的 所以需要把小数点后的替换为空 使其成为整数
#gsub函数将小数点后的替换为空后赋值给ENSEMBL
ENSEMBL<-gsub("\\.\\d*","",raw_count_filt$gene_id)
#将ENSEMBL重新添加到raw_count_filt矩阵 因为只是加了进去 并不是以矩阵的形式 所以加之后raw_count_filt的第一列依旧有小数点
row.names(raw_count_filt)<-ENSEMBL
#合并矩阵ENSEMBL和raw_count_filt 合并后看起来有七列 实则只有六列 最前面的一列是前面的步骤直接加进去的
raw_count_filt1<-cbind(ENSEMBL,raw_count_filt)
colnames(raw_count_filt1)<-c("ensembl_gene_id","gene_id","LoGlu_Rep1","LoGlu_Rep2","HiGlu_Rep1","HiGlu_Rep2")
注释
#对基因进行注释获取gene_symbol
#用bioMart对ensembl_id转换成gene_symbol
#安装以及载入包
install.packages('BiocManager')
BiocFileCache::install("biomaRt")
library('biomaRt')
library("curl")
#选择物种 mmusculus_gene_ensembl是老鼠 hsapiens_gene_ensembl是人
mart<-useDataset("mmusculus_gene_ensembl",useMart("ensembl"))
#待输入的基因ID是数据的行名
my_ensembl_gene_id<-row.names(raw_count_filt1)
提高连接时间
options(timeout=4000000)
#利用getBM函数获取gene_symbol 如果是小鼠用external_gene_name 人用hgnc_symbol
mms_symbols<-getBM(attributes=c('ensembl_gene_id',
'external_gene_name',
'chromosome_name',
'start_position',
'end_position',
'band'),
filters='ensembl_gene_id',
values=my_ensembl_gene_id,
mart=mart)
#将合并后的表达数据矩阵raw_count_filt1整合为一
readcount<-merge(raw_count_filt1,mms_symbols,by="ensembl_gene_id")
#网页版
#https://biodbnet-abcc.ncifcrf.gov/db/db2db.php 这个转换的不全 不知道原因
#https://www.biotools.fr/mouse/ensembl_symbol_converter 这个是全的 但是最后的一些数据没有
readcount<-read.table("D:/Users/hemiao/Desktop/shiyan/raw_count_filt.txt",sep="\t",header=TRUE)
#输出count文件
write.csv(readcount,file='readcount.csv')
查看readcount文件
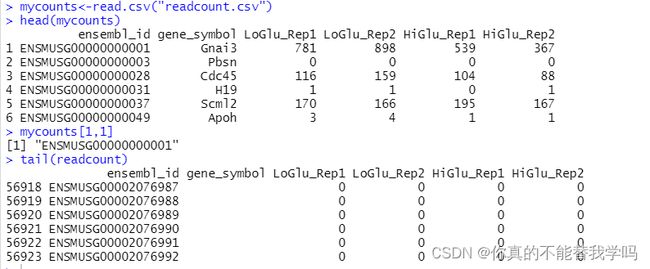
6 DEseq2筛选差异表达基因并用bioMart注释
6.1 DEseq2筛选差异表达基因
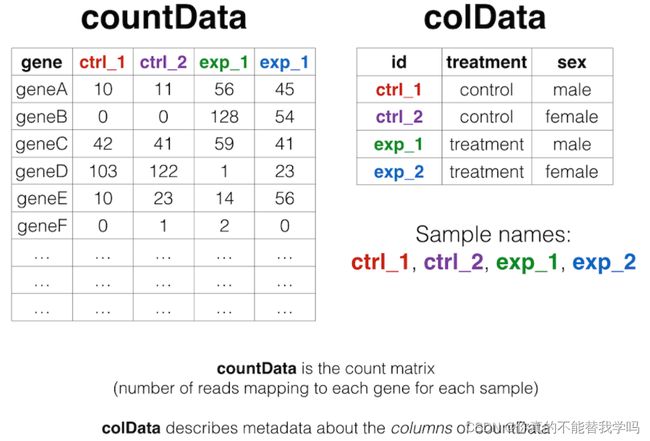
开始之前需要准备两个矩阵:
countData表示的是count矩阵,行代表gene,列代表样品,中间的数字代表对应count数;
colData表示sample的元数据,因为这个表提供了sample的元数据。
#1载入数据(countData和colData)
install.packages("tidyverse")
BiocManager::install("DESeq2")
library(tidyverse)
library(DESeq2)
#读取readcount.csv文件 文件在当前工作路径 所以没有写绝对地址 mycounts没有gene_symbol这一列 只有四列
mycounts<-read.csv("readcount.csv")
head(mycounts)
#多了一个x需要去除 先把第一列当作行名来处理 此时x那一列数值是空 就可以删除 行不属于列
rownames(mycounts)<-mycounts[,1]
mycounts<-mycounts[,-1]
mycounts<-mycounts[,-1]
#condition是因子 不是样本名称 小鼠数据有对照和处理组 各两个
condition<-factor(c(rep("LoGlu_Rep",2),rep("HiGlu_Rep",2)),levels=c("LoGlu_Rep","HiGlu_Rep"))
#coldData也可以自己在excel做好在导入
colData<-data.frame(row.names=colnames(mycounts),condition)
#2构建dds对象 开始DESeq流程
#dds=DESeqDataSet Object并标准化 对原始数据进行normalize
dds<-DESeqDataSetFromMatrix(mycounts,colData,design=~condition)
#进行差异表达分析
dds<-DESeq(dds)
dds
#3总体结果查看
#查看treat versus control的总体结果 并根据p-value进行重新排序 利用summary命令统计显示一共多少个genes上调和下调(FDR0.1)
#contrast参数有三个元素时依次代表数据中作为treat/control标签的列的名称 fold change的分子 以及fold change的分母 fold一般算的是treat/control(处理/控制)
res=results(dds,contrast=c("condition","LoGlu_Rep","HiGlu_Rep"))
res=res[order(res$pvalue),]
head(res)
summary(res)
#4提取差异表达基因(DEGs)并进行gene symbol注释
#查看符合阈值的差异基因有多少个 获取padj(p值经过多重校验校正后的值)小于0.05 表达倍数取以2为对数后大于1或者小于-1的差异表达基因
diff_gene_deseq2<-subset(res,padj<0.05&abs(log2FoldChange)>1)
diff_gene_deseq2<-subset(res,padj<0.05&(log2FoldChange>1|log2FoldChange<1))
#展示diff_gene_deseq2的维度和前几列
dim(diff_gene_deseq2)
head(diff_gene_deseq2)
write.csv(diff_gene_deseq2,file="DEG_treat_vs_control.csv")
查看mycounts和colData

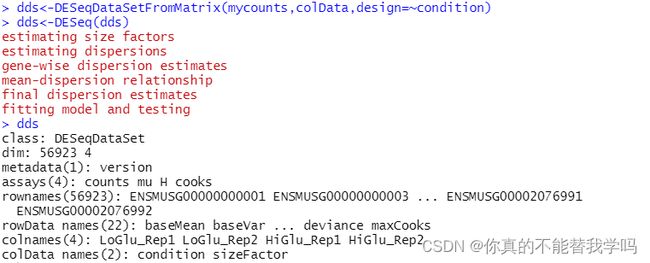
查看dds
能够看到dds的类型,维度,行名,colnames等等。
查看res
能够看到每个基因的pvalue,padj等信息,在summary中得知上调基因471个,下调基因468个,没有离群值。


查看diff_gene_seq2

6.2 用bioMart对差异表达基因进行注释
#5用bioMart对差异表达基因进行注释
library('biomaRt')
library("curl")
mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl"))
my_ensembl_gene_id<-row.names(diff_gene_deseq2)
mms_symbol<- getBM(attributes=c('ensembl_gene_id','external_gene_name',"description"),
filters = 'ensembl_gene_id', values = my_ensembl_gene_id, mart = mart)
head(mms_symbol)
#6合并数据 res结果和mms_symbol合并成一个文件
#查看数据后两个文件没有相同的列名 所以要给diff_gene_deseq2添加一个ensembl_gene_id的列名
#cbind增加新属性 也就是增加列 把原本的行名变成了列名
ensembl_gene_id<-rownames(diff_gene_deseq2)
diff_gene_deseq2<-cbind(ensembl_gene_id,diff_gene_deseq2)
colnames(diff_gene_deseq2)[1]<-c("ensembl_gene_id")
diff_name<-merge(diff_gene_deseq2,mms_symbol,by="ensembl_gene_id")
查看mms_symbol
mms_symbol和mms_symbols行名都是ensembl_gene_id,展示的内容不一样,前者的数据是后者的子集,经过padj筛选的。
注释后得到了每个ensembl_gene_id对应的gene_symbol也就是external_gene_name.

7 数据可视化 绘图
7.1 MA图
#没有经过statistical moderation平缓log2 fold changes的情况
#padj越小表达量越高
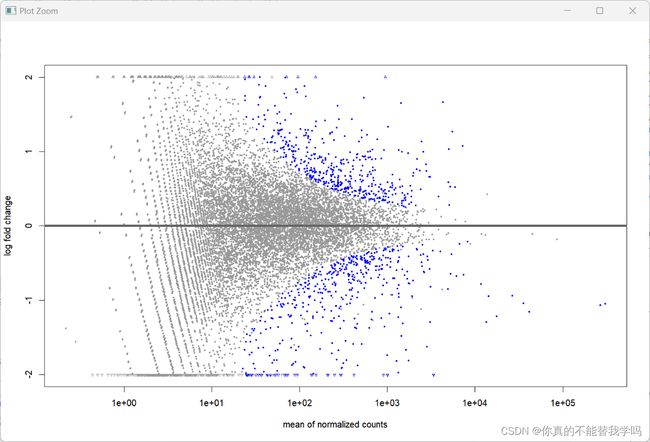
plotMA(res,ylim=c(-2,2))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=6, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
#经过lfcShrink收缩log2 fold change
#前面res结果已经按padj排序了 所以这次要按照行名升序再排列回来 否则和dds不一致
res_order<-res[order(row.names(res)),]
res = res_order
res.shrink <- lfcShrink(dds, contrast = c("condition","LoGlu_Rep","HiGlu_Rep"), res=res)
plotMA(res.shrink,ylim=c(-5,5))
#但结果提示Error in lfcShrink(dds, contrast = c("condition", "treat", "control"), : type='apeglm' shrinkage only for use with 'coef' 目前不确定原因 采取了下面的方式
#以apeglm包中的收缩估计量 成功
resApe <-lfcShrink(dds, coef=2,type="apeglm")
plotMA(resApe, ylim = c(-5,5),main="apeglm")
#以DESeq包中默认收缩估计值也成功 但是和Ape没什么区别
resLFC <- lfcShrink(dds, coef=2)
plotMA(resLFC, ylim = c(-5,5),main="normal")
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
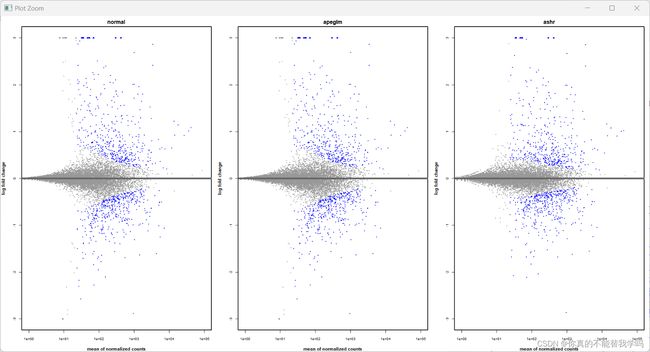
#比较三种收缩方式 Ash有差别
resApe <-lfcShrink(dds, coef=2,type="apeglm")
resAsh <-lfcShrink(dds, coef=2,type="ashr")
par(mfrow=c(1,3), mar=c(4,4,2,1))
xlim <- c(1,1e5); ylim<-c(-3,3)
plotMA(resLFC, xlim=xlim, ylim=ylim, main="normal")
plotMA(resApe, xlim=xlim, ylim=ylim, main="apeglm")
plotMA(resAsh, xlim=xlim, ylim=ylim, main="ashr")
par(opar)
查看MA图
MA 图可以用于展示数据表达是否异常的。MA 图上,一般是上下对称,离散的基因一般就是上调或者下调的基因,我觉得这个和看火山图很像。但是相较于火山图,用 MA判断整个数据的表达会更好,MA 图上,左侧大右侧小,某一个组织,某一个时期,少数基因高表达,多数基因低表达。这张图上,灰色的是正常表达基因,上方的蓝色是表达上调的基因,下方的蓝色是表达下调的基因。
7.2 Plot counts
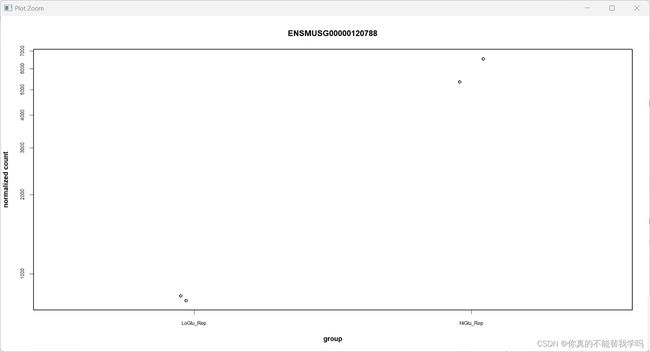
#DESeq2提供了一个plotCounts()函数来查看某一个感兴趣的gene在组间的差别 counts会根据groups分组
#不画图 只显示数据
plotCounts(dds, gene=which.min(res$padj), intgroup="condition", returnData=TRUE)
#只画图 不显示数据
plotCounts(dds, gene="ENSMUSG00000120788", intgroup="condition", returnData=FALSE)
7.3 PCA
vsdata<-vst(dds, blind=FALSE)
plotPCA(vsdata, intgroup="condition")

7.4 热图
更多使用方法可参考: https://blog.csdn.net/qq_35294674/article/details/122112145
library("pheatmap")
select<-order(rowMeans(counts(dds, normalized = TRUE)),
decreasing = TRUE)[1:20]
df <- as.data.frame(colData(dds)[,c("condition","sizeFactor")])
ntd <- normTransform(dds)
pheatmap(assay(ntd)[select,], cluster_rows=FALSE, show_rownames=FALSE,
cluster_cols=FALSE, annotation_col=df)

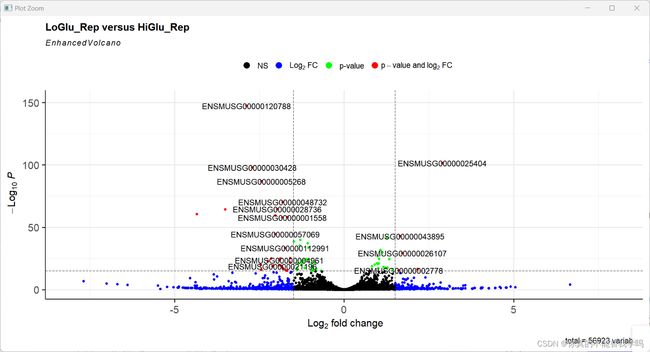
7.5 火山图
具体使用方法可参考: https://blog.csdn.net/qazplm12_3/article/details/93330560
BiocManager::install("EnhancedVolcano")
library(EnhancedVolcano)
EnhancedVolcano(res,
lab = rownames(res),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-8, 8),
title = 'LoGlu_Rep versus HiGlu_Rep',
pCutoff = 10e-16,
FCcutoff = 1.5,
col=c('black','blue','green','red1'),
colAlpha=1,
)
8 富集分析 功能注释
9 KEGG通路可视化
以上大部分参考这一篇文章: https://www.jianshu.com/p/e8cd62ba14fe