AlphaFold2源码解析(3)--数据预处理
AlphaFold2源码解析(3)–数据预处理
数据预处理整体流程

数据处理入口:
feature_dict = data_pipeline.process( input_fasta_path=fasta_path,# 输入序列目录 msa_output_dir=msa_output_dir) # MSA序列目录 可能是单体也可能是多聚体
主要调动的API是:
- 通过使用
run_msa_tool()函数调用jackhmer和hhblits对输入的Fasta文件进行MSA搜索,输出的格式为sto; template_searcher()使用去冗余后的MSA序列对pdb70数据库进行搜索,得到TemplateHits;template_featurizer()使用kalign程序对Hits的序列与input序列进行比对,得到模板信息;- 最后通过
make_*_features()函数对特征进一步处理,与融合得到alphafold所需的所有特征字典。
预处理工具
从上文我们知道,Alphafold需要与专家经验库进行交互,抽取MSA特征。这里用到了多个生物学工具:
- openmm:用于分析模拟的高性能工具包,是 Omnia(预测生物分子模拟的工具套件)的一部分。
- kalign:一款针对大规模基因组序列的多序列对比工具,运行速度与准确度都比较高。
- jackhmmer:一款高效的蛋白质搜索工具,用于从序列数据库中,找到同源的序列。
- pdbfixer:用于修复PDB数据,对其中蛋白质结构进行基本的准备或者修复。
- HHsuite:一款蛋白质研究软件,快速搜索分析蛋白质的功能特性。
jackhmmer
ackhmmer针对seqdb中的目标序列迭代搜索seqfile中的每个查询序列。第一次迭代与phmmer搜索相同。对于下一次迭代,将查询的多重比对与满足包含阈值的所有目标序列组合在一起,从该比对构建配置文件(与在比对上使用hmmbuild相同),并完成seqdb的配置文件搜索(与带有配置文件的hmmsearch)。
查询seqfile可以是“-”(破折号),在这种情况下,查询序列是从标准输入管道而不是从文件中读取的。无法从标准输入流中读取 seqdb,因为jackhmmer需要对数据库进行多次传递。
输出格式被设计成人类可读的,但往往过于庞大以至于阅读起来不切实际,解析起来也很痛苦。–tblout和–domtblout选项以简洁且易于解析的简单表格格式保存输出。-o选项允许重定向主要输出,包括将其丢弃在 /dev/null 中。
参数详情可以参考:https://www.mankier.com/1/jackhmmer
在alphaFold2的代码路径
有几个数据库使用了jackhmmer来搜索MSA,如 uniref90、mgnify
./alphafold/data/tools/jackhmmer.py
def _query_chunk(self,
input_fasta_path: str,
database_path: str,
max_sequences: Optional[int] = None) -> Mapping[str, Any]:
"""Queries the database chunk using Jackhmmer."""
with utils.tmpdir_manager() as query_tmp_dir:
sto_path = os.path.join(query_tmp_dir, 'output.sto')
# The F1/F2/F3 are the expected proportion to pass each of the filtering
# stages (which get progressively more expensive), reducing these
# speeds up the pipeline at the expensive of sensitivity. They are
# currently set very low to make querying Mgnify run in a reasonable
# amount of time.
cmd_flags = [
# Don't pollute stdout with Jackhmmer output.
'-o', '/dev/null', # https://www.mankier.com/1/jackhmmer /dev/null 系统自带的不保存文件
'-A', sto_path, # 在最终迭代之后,将所有命中的带注释的多个对齐方式保存满足包含阈值(还包括原始查询)的多个对齐,以在Stockholm格式中。
'--noali', # 在最终迭代之后,将所有命中的带注释的多个对齐方式保存满足包含阈值(还包括原始查询)的多个对齐,以在Stockholm格式中。
'--F1', str(self.filter_f1), # 第一个滤波器阈值;为MSV滤波器步骤设置P值阈值。默认值为0.02,这意味着预计最高得分的非同源目标的2%将通过过滤器。
'--F2', str(self.filter_f2), # 第二滤波阈值;设置维特比滤波步骤的p值阈值。默认值是0.001
'--F3', str(self.filter_f3), # 第三滤波器阈值;为正滤波器步骤设置p值阈值。默认值为1E-5。
'--incE', str(self.e_value), # 包括e值<= 的序列在后续迭代或由-A输出的最终对齐中。默认值是0.001。
# Report only sequences with E-values <= x in per-sequence output.
'-E', str(self.e_value),
'--cpu', str(self.n_cpu),
'-N', str(self.n_iter)
]
if self.get_tblout:
tblout_path = os.path.join(query_tmp_dir, 'tblout.txt') # 在最后的迭代之后,将最高序列命中的表格摘要保存到中
cmd_flags.extend(['--tblout', tblout_path])
if self.z_value: # 声明数据库的总大小为序列,以用于e值计算。通常情况下,e值是根据实际搜索的数据库大小计算的(例如,目标seqdb中的序列数量)。在某些情况下(例如,如果您已将目标序列数据库拆分为多个文件以并行化搜索),您可能更了解搜索空间的实际大小。
cmd_flags.extend(['-Z', str(self.z_value)])
if self.dom_e is not None:
cmd_flags.extend(['--domE', str(self.dom_e)]) # 报告每个域输出中条件e值<= 的域,以及每个显著序列命中得分最高的域。默认值是10.0。
if self.incdom_e is not None:
cmd_flags.extend(['--incdomE', str(self.incdom_e)]) #在后续迭代或由-A输出的最终对齐输出中包括条件e值<= 的域,除了每个显著序列命中的得分最高的域。默认值是0.001。
cmd = [self.binary_path] + cmd_flags + [input_fasta_path,
database_path]
logging.info('Launching subprocess "%s"', ' '.join(cmd))
process = subprocess.Popen(
cmd, stdout=subprocess.PIPE, stderr=subprocess.PIPE)
with utils.timing(
f'Jackhmmer ({os.path.basename(database_path)}) query'):
_, stderr = process.communicate()
retcode = process.wait()
if retcode:
raise RuntimeError(
'Jackhmmer failed\nstderr:\n%s\n' % stderr.decode('utf-8'))
# Get e-values for each target name
tbl = ''
if self.get_tblout:
with open(tblout_path) as f:
tbl = f.read()
if max_sequences is None:
with open(sto_path) as f:
sto = f.read()
else:
sto = parsers.truncate_stockholm_msa(sto_path, max_sequences)
raw_output = dict(
sto=sto,
tbl=tbl,
stderr=stderr,
n_iter=self.n_iter,
e_value=self.e_value)
return raw_output
HHsuite
HHsuite 工具简要教程
hhblits:迭代地使用查询序列或 MSA 搜索 HHsuite 数据库
hhsearch:使用查询 MSA 或 HMM 搜索 HHsuite 数据库
hhmake:从输入 MSA 构建 HMM
hhfilter:按最大序列标识、覆盖率和其他标准过滤 MSA
hhalign:计算两个 HMM/MSA 的成对对齐等
hhconsensus:计算 A3M/FASTA 输入文件的一致序列
reformat.pl:重新格式化一个或多个 MSA
adds.pl:将 PSIPRED 预测的二级结构添加到 MSA 或 HHM 文件
hhmakemodel.pl:从 HHsearch 或 HHblits 结果生成 MSA 或粗略 3D 模型
hhmakemodel.py:从 HHsearch 或 HHblits 结果生成粗略的 3D 模型并修改 cif 文件以使其与 MODELLER 兼容
hhsuitedb.py:使用预过滤、打包的 MSA/HMM 和索引文件构建 HHsuite 数据库
splitfasta.pl : 将一个多序列 FASTA 文件拆分成多个单序列文件
renumberpdb.pl:生成 PDB 文件,索引重新编号以匹配输入序列索引
HHPaths.pm:带有 PDB、BLAST、PSIPRED 等路径的配置文件。
mergeali.pl:根据种子序列的 MSA 以 A3M 格式合并 MSA
pdb2fasta.pl:从全局 pdb 文件的 SEQRES 记录生成 FASTA 序列文件
cif2fasta.py:从 globbed cif 文件的 entity_poly 的 pdbx_seq_one_letter_code 条目生成 FASTA 序列
pdbfilter.pl:从 pdb2fasta.pl 输出中生成一组具有代表性的 PDB/SCOP 序列
pdbfilter.py:从 cif2fasta.py 输出生成一组具有代表性的 PDB/SCOP 序列
调用不带参数或带-h选项的程序以获得更详细的解释。
具体参数参考:https://github.com/soedinglab/hh-suite/wiki#hhsearch–search-a-database-of-hmms-with-a-query-msa-or-hmm
HHSearch
AlphaFold 主要用到的地方时模版搜索,使用的是HHSearch在uniref90中搜索同源模版
alphafold/data/tools/hhsearch.py
## 入口
pdb_template_hits = self.template_searcher.get_template_hits(
output_string=pdb_templates_result, input_sequence=input_sequence)
.....
## hhsearch d8li/output.hhr -maxseq 1000000 -d /data2/datasets/pdb70/pdb70
def query(self, a3m: str) -> str:
"""Queries the database using HHsearch using a given a3m."""
with utils.tmpdir_manager() as query_tmp_dir:
input_path = os.path.join(query_tmp_dir, 'query.a3m')
hhr_path = os.path.join(query_tmp_dir, 'output.hhr')
with open(input_path, 'w') as f:
f.write(a3m)
db_cmd = []
for db_path in self.databases:# 使用的数据库
db_cmd.append('-d')
db_cmd.append(db_path)
cmd = [self.binary_path,
'-i', input_path,# 输出
'-o', hhr_path, # 输出
'-maxseq', str(self.maxseq) # 最大序列个数
] + db_cmd
logging.info('Launching subprocess "%s"', ' '.join(cmd))
process = subprocess.Popen(
cmd, stdout=subprocess.PIPE, stderr=subprocess.PIPE)
with utils.timing('HHsearch query'):
stdout, stderr = process.communicate()
retcode = process.wait()
if retcode:
# Stderr is truncated to prevent proto size errors in Beam.
raise RuntimeError(
'HHSearch failed:\nstdout:\n%s\n\nstderr:\n%s\n' % (
stdout.decode('utf-8'), stderr[:100_000].decode('utf-8')))
with open(hhr_path) as f:
hhr = f.read()
return hhr
hhblits
与hhsearch类似, 区别HHblits 的运行速度比 HHsearch 快 30 到 3000 倍,但灵敏度仅降低了几个百分点。(这里使用的原因是bdf的数据比uniref90大,加快搜索速度)
def query(self, input_fasta_path: str) -> List[Mapping[str, Any]]:
"""Queries the database using HHblits."""
with utils.tmpdir_manager() as query_tmp_dir:
a3m_path = os.path.join(query_tmp_dir, 'output.a3m')
db_cmd = []
for db_path in self.databases:
db_cmd.append('-d')
db_cmd.append(db_path)
cmd = [
self.binary_path,
'-i', input_fasta_path,
'-cpu', str(self.n_cpu),
'-oa3m', a3m_path,
'-o', '/dev/null',
'-n', str(self.n_iter),
'-e', str(self.e_value),
'-maxseq', str(self.maxseq),
'-realign_max', str(self.realign_max),
'-maxfilt', str(self.maxfilt),
'-min_prefilter_hits', str(self.min_prefilter_hits)]
if self.all_seqs:
cmd += ['-all']
if self.alt:
cmd += ['-alt', str(self.alt)]
if self.p != _HHBLITS_DEFAULT_P:
cmd += ['-p', str(self.p)]
if self.z != _HHBLITS_DEFAULT_Z:
cmd += ['-Z', str(self.z)]
cmd += db_cmd
logging.info('Launching subprocess "%s"', ' '.join(cmd))
process = subprocess.Popen(
cmd, stdout=subprocess.PIPE, stderr=subprocess.PIPE)
with utils.timing('HHblits query'):
stdout, stderr = process.communicate()
retcode = process.wait()
if retcode:
# Logs have a 15k character limit, so log HHblits error line by line.
logging.error('HHblits failed. HHblits stderr begin:')
for error_line in stderr.decode('utf-8').splitlines():
if error_line.strip():
logging.error(error_line.strip())
logging.error('HHblits stderr end')
raise RuntimeError('HHblits failed\nstdout:\n%s\n\nstderr:\n%s\n' % (
stdout.decode('utf-8'), stderr[:500_000].decode('utf-8')))
with open(a3m_path) as f:
a3m = f.read()
raw_output = dict(
a3m=a3m,
output=stdout,
stderr=stderr,
n_iter=self.n_iter,
e_value=self.e_value)
return [raw_output]
kalign
之前提到的clustalo, muscle, mafft 适用于几千到几万条序列的多序列比对,在比较基因组学的分析中,需要对不同基因组的序列进行多序列比对。对于基因组规模的多序列比对而言,之前的工具运行速度上就不够理想了。
kalign 是一款针对大规模序列的多序列比对工具,无论是运行速度,还是比对的准确度,都令人满意。
Kalign 期望输入是一组 fasta 格式的未对齐序列或对齐的 fasta、MSF 或 clustal 格式的对齐序列。如果序列已经对齐,kalign 将删除所有间隙字符并重新对齐序列。
默认情况下,Kalign 会自动检测输入序列是蛋白质还是 DNA,并选择合适的比对参数。
详细参数请参考: https://github.com/TimoLassmann/kalign
alphafold/data/tools/kalign.py
def align(self, sequences: Sequence[str]) -> str:
logging.info('Aligning %d sequences', len(sequences))
for s in sequences:
if len(s) < 6:
raise ValueError('Kalign requires all sequences to be at least 6 '
'residues long. Got %s (%d residues).' % (s, len(s)))
with utils.tmpdir_manager() as query_tmp_dir:
input_fasta_path = os.path.join(query_tmp_dir, 'input.fasta')
output_a3m_path = os.path.join(query_tmp_dir, 'output.a3m')
with open(input_fasta_path, 'w') as f:
f.write(_to_a3m(sequences))
cmd = [
self.binary_path,
'-i', input_fasta_path,
'-o', output_a3m_path,
'-format', 'fasta',
]
logging.info('Launching subprocess "%s"', ' '.join(cmd))
process = subprocess.Popen(cmd, stdout=subprocess.PIPE,
stderr=subprocess.PIPE)
with utils.timing('Kalign query'):
stdout, stderr = process.communicate()
retcode = process.wait()
logging.info('Kalign stdout:\n%s\n\nstderr:\n%s\n',
stdout.decode('utf-8'), stderr.decode('utf-8'))
if retcode:
raise RuntimeError('Kalign failed\nstdout:\n%s\n\nstderr:\n%s\n'
% (stdout.decode('utf-8'), stderr.decode('utf-8')))
with open(output_a3m_path) as f:
a3m = f.read()
return a3m
预处理格式(包括输出和中间结果)
MSA 特征提取
bfd_uniclust_hits.a3m主要是通过uniclust30_2018_08和bfd_metaclust_clu_complete_id30_c90_final_seq两个数据集通过HHBlits构建

uniref90_hits.sto通过uniref90数据集Jackhmmer搜索得到

mgnify_hits.sto通过uniref90数据集mgnify搜索得到

构建MSA特征入口:
msa_features = make_msa_features((uniref90_msa, bfd_msa, mgnify_msa)) # 构造MSA特征
对MSA特征去重、MSA序列数字list化以及更新特征:
def make_msa_features(msas: Sequence[parsers.Msa]) -> FeatureDict:
"""Constructs a feature dict of MSA features."""
if not msas:
raise ValueError('At least one MSA must be provided.')
int_msa = []
deletion_matrix = []
species_ids = []
seen_sequences = set()
for msa_index, msa in enumerate(msas):
if not msa:
raise ValueError(f'MSA {msa_index} must contain at least one sequence.')
for sequence_index, sequence in enumerate(msa.sequences):
if sequence in seen_sequences:
continue
seen_sequences.add(sequence)
int_msa.append(
[residue_constants.HHBLITS_AA_TO_ID[res] for res in sequence])
deletion_matrix.append(msa.deletion_matrix[sequence_index])
identifiers = msa_identifiers.get_identifiers(
msa.descriptions[sequence_index])
species_ids.append(identifiers.species_id.encode('utf-8'))
num_res = len(msas[0].sequences[0])
num_alignments = len(int_msa)
features = {}
features['deletion_matrix_int'] = np.array(deletion_matrix, dtype=np.int32)
features['msa'] = np.array(int_msa, dtype=np.int32)
features['num_alignments'] = np.array(
[num_alignments] * num_res, dtype=np.int32)
features['msa_species_identifiers'] = np.array(species_ids, dtype=np.object_)
return features
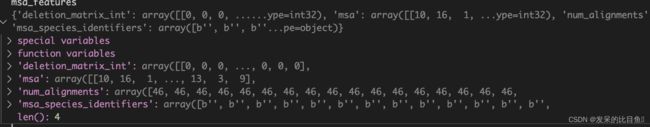
MSA特征字典:
deletion_matrix_int:检测MSA中每条序列中是否存在小写字符的氨基酸信息,此区域代表同源序列中的序列被删除。(在推理中貌似没用,矩阵的维度是NxL),可能的信息Training时在Residue cropping部分。msa:将MSA每条序列转为以数字替代的list,方便后续one-hot化num_alignments:对齐数量msa_species_identifiers:种属信息,貌似我提供的MSA中没有这部分东西
Template特征提取
pdb_hits.hhr 是uniref90MSA通过HHSearch检索pdb70库得到的信息, 其中主要记录的是可用模板的PDB ID,序列相似度,结构与序列match的区间, 在HHSearch运行中,唯一设置为非默认值的标志是-maxseq 1000000。每个HIT的结构数据都是从PDB数据库中相应的mmCIF文件中获得。如果PDB70中的序列与mmCIF文件中的序列不完全匹配,则使用Kalig对两者进行比对。
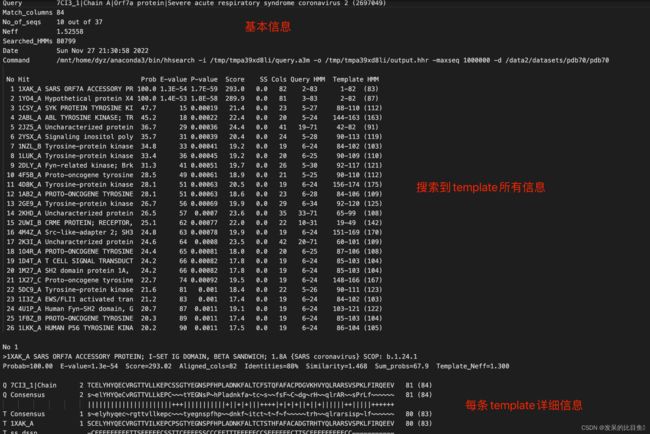
读取.hhr文件信息
pdb_template_hits = self.template_searcher.get_template_hits(output_string=pdb_templates_result, input_sequence=input_sequence)
@dataclasses.dataclass(frozen=True)
class TemplateHit:
"""Class representing a template hit."""
index: int
name: str
aligned_cols: int
sum_probs: Optional[float]
query: str
hit_sequence: str
indices_query: List[int]
indices_hit: List[int]
aligned_cols:指query sequence被模板覆盖的区间大小sum_probs被用于对template进行打分排名indices_query,indices_hit是最重要的信息,这里记录了模板和query sequence的匹配区间信息
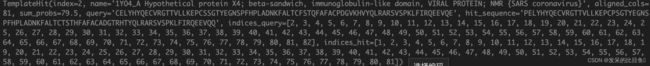
template_hits 特征化
TEMPLATE_FEATURES = {
'template_aatype': np.float32,
'template_all_atom_masks': np.float32,
'template_all_atom_positions': np.float32, #对template atom 位置信息
'template_domain_names': np.object, # 领域
'template_sequence': np.object, # template序列
'template_sum_probs': np.float32, # 对template进行打分排名
}
def get_templates(
self,
query_sequence: str,
hits: Sequence[parsers.TemplateHit]) -> TemplateSearchResult:
"""Computes the templates for given query sequence (more details above). 计算给定查询序列的模板(以上详细信息)。"""
logging.info('Searching for template for: %s', query_sequence)
........
result = _process_single_hit(
query_sequence=query_sequence,
hit=hit,
mmcif_dir=self._mmcif_dir,# pdb mmcif文件地址
max_template_date=self._max_template_date, # 模板日期
release_dates=self._release_dates,
obsolete_pdbs=self._obsolete_pdbs,
strict_error_check=self._strict_error_check,
kalign_binary_path=self._kalign_binary_path)
........
return TemplateSearchResult(
features=template_features, errors=errors, warnings=warnings)
.....
def _extract_template_features(....)
.......
return (
{
'template_all_atom_positions': np.array(templates_all_atom_positions),
'template_all_atom_masks': np.array(templates_all_atom_masks),
'template_sequence': output_templates_sequence.encode(),
'template_aatype': np.array(templates_aatype),
'template_domain_names': f'{pdb_id.lower()}_{chain_id}'.encode(),
}

template的feature特征包括: N N N指模板的数量
template_aatype: 模板序列的one-hot representation,shape = NxLx22,包括unknown和gap;template_all_atom_masks: shape = NxLx37,代表在模板中,原子是否存在,存在=1,不存在=0;template_all_atom_positions: shape = Lx37x3, 其中37为所有的可能的蛋白原子类型,3维代表xyz坐标值。template_domain_names: 模板的名称template_sequence: shape =NxL 序列字符串template_sum_probs: match的打分值 (np.float32)
最后返回的时TemplateSearchResult实例。
input sequence特征提取
这里主要是对输入序列进行特征化:
### 入口
sequence_features = make_sequence_features(
sequence=input_sequence,
description=input_description,
num_res=num_res)## 构造输入序列特征
def make_sequence_features(
sequence: str, description: str, num_res: int) -> FeatureDict:
"""Constructs a feature dict of sequence features. 构造序列特征的特征字典。"""
features = {}
features['aatype'] = residue_constants.sequence_to_onehot(
sequence=sequence,
mapping=residue_constants.restype_order_with_x,
map_unknown_to_x=True)#残基one-hot
features['between_segment_residues'] = np.zeros((num_res,), dtype=np.int32)
features['domain_name'] = np.array([description.encode('utf-8')],
dtype=np.object_)
features['residue_index'] = np.array(range(num_res), dtype=np.int32)
features['seq_length'] = np.array([num_res] * num_res, dtype=np.int32)
features['sequence'] = np.array([sequence.encode('utf-8')], dtype=np.object_)
return features
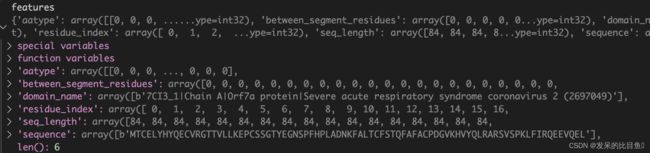
特征字典信息:
aatype: one-hot编码的序列between_segment_residues?含义不明(可能在multimer的文章中会有解释)domain_name:序列名,字符串信息residue_index: 残基编号,从0开始seq_length:序列长度,为什么会重复N次?sequence:人类可读的序列3字母缩写序列,类型字符串
参考
https://zhuanlan.zhihu.com/p/492381344