LncFinder | 非编码RNA的识别与分析神器!!!~

1写在前面
非编码RNA(ncRNAs), 是指不编码蛋白质的RNA。
其中包括rRNA,tRNA,snRNA,snoRNA, lncRNA和miRNA等多种已知功能的RNA,还包括未知功能的RNA。
长链非编码RNA(lncRNA)指的是长于200核苷酸的不编码蛋白质的转录物(随着对ncRNAs的不对认知,这个概念也在不断修改)。
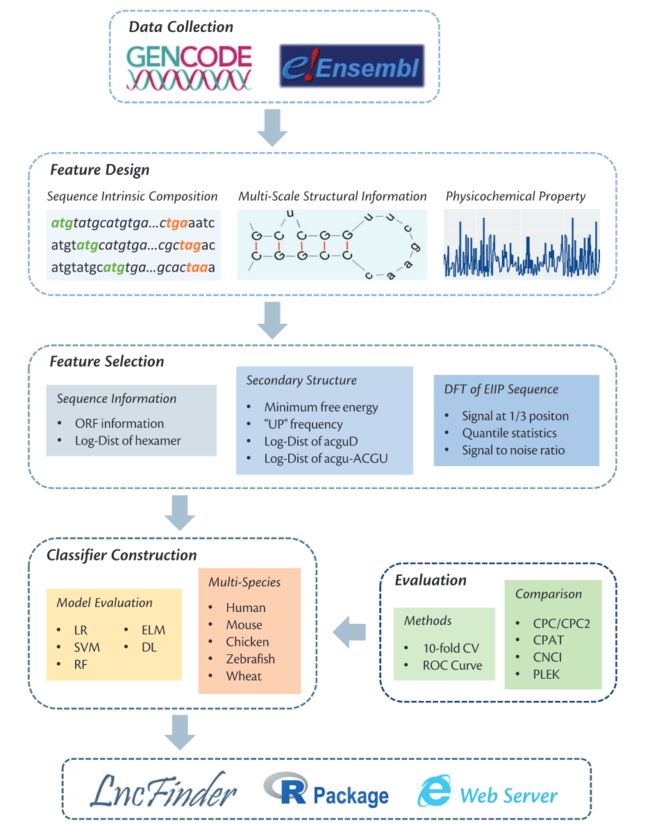
在我们完成测序后,一个很重要的步骤就是识别序列,本期我们介绍一个基于Logarithm-Distance, Multi-Scale Structural Information和EIIP-Based Physicochemical Property的lncRNA识别与分析工具,LncFinder。
2用到的包
rm(list = ls())
library(LncFinder)
library(tidyverse)
3示例数据
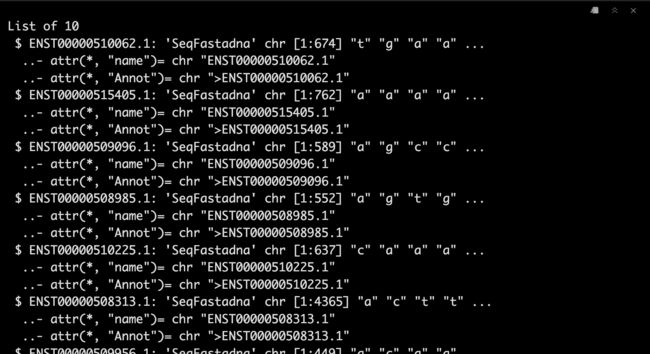
包内提供了示例数据,我们一起看一下吧。
data("demo_DNA.seq")
Seqs <- demo_DNA.seq
str(Seqs)
4不基于二级结构特征的lncRNA预测
4.1 开始预测
result_1 <- LncFinder::lnc_finder(Seqs, #fasta或二级结构seq
SS.features = F, ## 二级结构seq
format = "DNA", ## 'DNA' / 'SS'
frequencies.file = "human",
svm.model = "human",
parallel.cores = 2 ## -1为调用所有cores
)
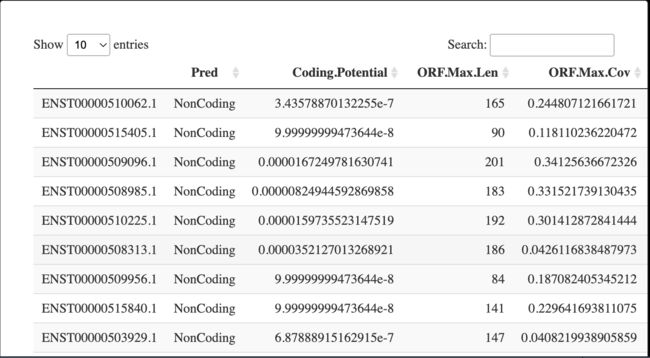
4.2 查看结果
DT::datatable(result_1)
5基于二级结构特征的lncRNA预测
这里我们就需要有一个二级结构序列文件(Dot-Bracket Notation)做为input了。
5.1 windows下运行
## 转为string
Seqs_windows <- sapply(Seqs, seqinr::getSequence, as.string = T)
## 写为fasta文件
seqinr::write.fasta(Seqs_windows,
names = names(Seqs_windows),
file.out = "tmp.RNA.fa",
as.string = T
)
RNAfold.path <- '"E:/Program Files/ViennaRNA/RNAfold.exe"'
RNAfold.command <- paste(RNAfold.path, "--noPS -i tmp.RNA.fa -o output")
system(RNAfold.command)
5.2 UNIX/Linux下运行
data("demo_DNA.seq")
Seqs_unix <- demo_DNA.seq
Seqs_unix <- LncFinder::run_RNAfold(Seqs_unix,
RNAfold.path = "RNAfold",
parallel.cores = 2)
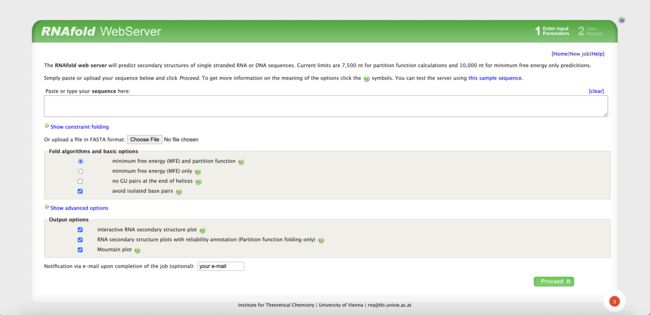
5.3 网页获取
网址如下:
http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi
5.4 开始预测
result_2 <- LncFinder::lnc_finder(Seqs_unix,
SS.features = T,
format = "SS",
frequencies.file = "human",
svm.model = "human",
parallel.cores = 4)
5.5 查看结果
这里我们就可以获得.和()组成的Dot-Bracket了。
DT::datatable(result_2)

6构建新模型
6.1 创建frequency文件
这里输入文件是DNA序列。
data(demo_DNA.seq)
Seqs <- demo_DNA.seq
myFile_1 <- LncFinder::make_frequencies(cds.seq = Seqs[1:5],
lncRNA.seq = Seqs[5:10],
SS.features = F,
cds.format = "DNA",
lnc.format = "DNA",
check.cds = T,
ignore.illegal = F
)
当然你的input也可以是前面计算好的二级结构文件。
data(demo_SS.seq)
SS.Seq <- demo_SS.seq
myFile_2 <- LncFinder::make_frequencies(cds.seq = SS.Seq,
lncRNA.seq = Seqs,
SS.features = F,
cds.format = "SS",
lnc.format = "DNA",
check.cds = F,
ignore.illegal = T)
我们甚至可以再复杂一点,把这两个示例结果都用上。
myFile_3 <- LncFinder::make_frequencies(cds.seq = Seqs,
mRNA.seq = SS.Seq[1:5],
lncRNA.seq = SS.Seq[6:10],
SS.features = T,
cds.format = "DNA",
lnc.format = "SS",
check.cds = T,
ignore.illegal = T)
6.2 开始构建新模型
这里为了节约时间,我们把range设置的小一点,大家在实际应用中使用默认值就好了。
myFile <- LncFinder::make_frequencies(cds.seq = Seqs,
mRNA.seq = SS.Seq[1:5],
lncRNA.seq = SS.Seq[6:10],
SS.features = T,
cds.format = "DNA",
lnc.format = "SS",
check.cds = T,
ignore.illegal = F)
myModel <- LncFinder::build_model(mRNA.seq = SS.Seq[1:5],
lncRNA.seq = SS.Seq[6:10],
frequencies.file = myFile,
SS.features = TRUE,
lncRNA.format = "SS",
mRNA.format = "SS",
parallel.cores = -1,
folds.num = 10,
seed = 123,
gamma.range =
(2^seq(-2, 0, 1)), ## ranges for SVM parameter
cost.range = c(1, 4, 8) ## the ranges for SVM parameter
)
6.3 pre-built model
这个包里包含了一些预制好的model,大家有兴趣可以在这个网址看到:
https://github.com/HAN-Siyu/LncFinder/tree/master/Data/Additional%20Models
然后通过load使用吧。
load("gallus.data.RData")
7调整SVM模型参数
这里我们调用一下已经识别的示例数据。
data("demo_dataset")
dat <- demo_dataset
DT::datatable(dat)
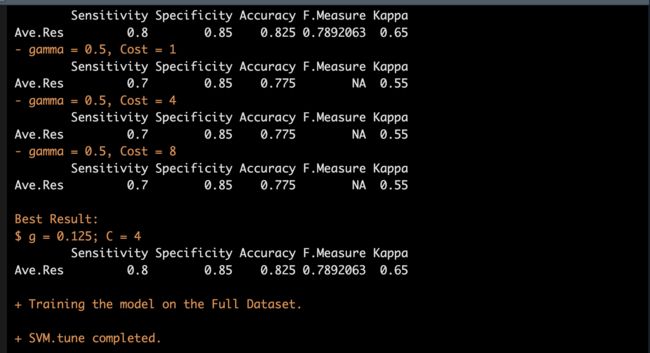
接着我们将return.model设为T,返回最佳模型。
同样这里为了节约时间,我们把range设置的小一点。
拿到最佳模型就开始去identify吧。
tuneSVM <- LncFinder::svm_tune(dataset = dat,
positive.class = "NonCoding",
folds.num = 5,
seed = 123,
gamma.range = (2^c(-3, -1)),
cost.range = c(1, 4, 8),
return.model = T,
parallel.cores = -1)
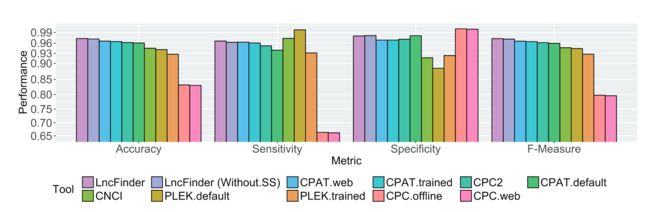
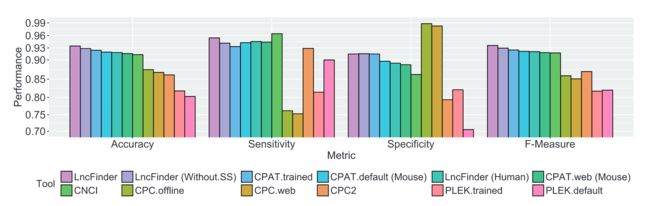
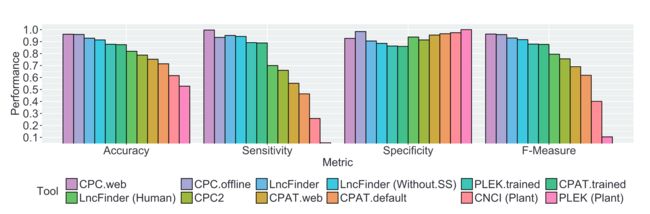
8LncFinder的表现
按照原文中的比较,在人类、小鼠、小麦数据集上,表现都比较出色, 优于其他方法(左右滑动查看吧):
9如何引用
Han S, Liang Y, Ma Q, et al. LncFinder: an integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief Bioinform. 2019;20(6):2009-2027. doi:10.1093/bib/bby065

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
ComplexHeatmap | 颜狗写的高颜值热图代码!
ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
scRNA-seq | 吐血整理的单细胞入门教程
NetworkD3 | 让我们一起画个动态的桑基图吧~
RColorBrewer | 再多的配色也能轻松搞定!~
rms | 批量完成你的线性回归
CMplot | 完美复刻Nature上的曼哈顿图
Network | 高颜值动态网络可视化工具
boxjitter | 完美复刻Nature上的高颜值统计图
linkET | 完美解决ggcor安装失败方案(附教程)
......

本文由 mdnice 多平台发布