测序数据量那些事
上周同事问了我集群存储大小、测序数据量之间的关系,我只是回复集群存储大小记录的是计算机的存储单位,测序数据量是碱基的测序个数。至于两者之间的换算逻辑,因果逻辑推算讲解的不是太明白,借用这篇文章给与捋顺。
1、计算机存储与测序数据量的关系
两者之间没有直接关系,但两者之间存在着因果关系。
测序数据量描述的是一个 巨大的 文本文件,这个文件里面含有若干个A、T、C、G字母组成。在测序语境下,我们把A、T、C、G为 碱基(bp,base pair),也即 测序数据量描述的是一个 由若干个碱基(bp)组成的文本文件。
计算机存储描述的是 文本文件在计算机环境下的占用的存储空间大小。换算思路为,碱基 A => ASCII 码 => 二进制 01 => 计算机最基本的储存单元比特 bit 。
1个碱基A(bp) ,以ASCII码存储,占用了2个字节(2B)的计算机存储空间,可参考下图:
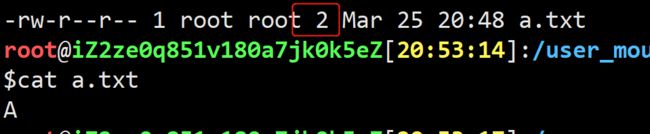
备注:网上说1个A,以ASCII码存储,占用的存储为1字节(1B),但我实际看到的是2B,有知道的可以在评论区为我解惑。2.2部分 还是按照网上说的1碱基文件文件 1B来计算。
2、人类基因组有多大
问题:人体基因组共有30亿个碱基对,其在集群中占据的计算机存储空间有多少?
计算前基础知识:
① 人类基因组只记录单链30亿个碱基信息,另一个链信息通过碱基互补配对推算;
计算一个含有30亿个碱基(bp)信息的文本文件即可,无需考虑双链信息(即无需X2);
② 30亿个碱基;
意思是" 一行特别长的字,共有30亿个,每个字恰好是A、T、G、C中的一个,恰好放在了一个文本文件中";
③ 计算机存储换算单位
1个碱基(A/T/C/G,bp)占用的存储空间为2比特(bit),8bit(比特) = 1B(字节);
常用换算单位还有: 8bit=1Byte;1024B=1KB(KiloByte);1024KB=1MB(MegaByte);1024MB=1GB(GigaByte);1024GB=1TB(TeraByte)
此时的KB、MB、GB、TB就是我们在集群上使用 “ls -l” 命令看到的文件大小的单位。
④ 碱基个数之间的换算单位
此时的换算单位为 1000 bp(base pair)= 1 Kb;1000 Kb = 1 Mb;1000 Mb=1 Gb;1000 Gb = 1 Tb。
2.1 按计算机二进制记录时:750MB
因为一共只有 ATCG 四种情况(可以转换成计算机的 00、01、10、11),所以每一个碱基,要用 2 位二进制(2 个 bits)记录。
30亿 x 2 = 60亿bits。
然后就是单位换算。计算机里,我们通常说的 KB、MB、GB 都是指“大B”, 1B = 8bits。
60亿bits / 8 = 750,000,000 B = 750 MB
此时的 750MB,我们读作“750M ”, “750兆”。
2.2 按纯文本记录时:3GB
30亿个碱基,我们读作 “3 G 碱基”,“3G basepair”。
上面二进制的文件,机器能读,而且体积小,传输用它就可以了,但人是不方便阅读的。
人能读的是 ASCII 码,是直接记录“A”、“T”、“C”、“G”这样的字符。一个 ASCII 字符,大小是 1B。
所以,如果按纯文本保存 30亿 个字母,30亿字母 = 3,000,000,000 B = 3 GB
此时的 3 GB,GB是计算机的文件大小GigaByte,可以读作“3G”;
2.3 人类基因组的长度
对于人类全基因组来说,长度大约3Gbp(Giga-basepairs)。
3,234.83 Mb (Mega-basepairs) per haploid genome
对于人类外显子组来说,长度大约是30Mbp(Mega-basepairs)
The exome of the human genome consists of roughly 180,000 exons constituting about 1% of the total genome, or about 30 megabases of DNA
3、reads、fastq、fastq.gz大小关系
随机取1000条reads,分别看其在文本格式(txt),文本格式(fastq),压缩文件(fastq.gz)的大小。
① reads(读长),指的是测序仪单次测序所得到的碱基序列;
② fastq,格式是一种保存生物序列(通常为核酸序列)及其测序质量得分信息的文本格式;
③fastq.gz,一般的fastq格式压缩后呈现的格式;
3.1 reads基本单位介绍
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;

第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
3.2 随机1000条PE150测序reads的大小(bp)
碱基数 = reads数 X 测序长度 X 双链,此时的总碱基数量为 1000 x 150 x 2 = 300,000 bp = 0.3 Mb 。读作0.3M个 碱基。
3.3 随机1000条PE150测序reads的大小(txt)
占用了计算机 367708字节(B)的存储空间,换算为360 KB。

3.4 随机1000条PE150测序reads的大小(fastq)
占用了计算机 367708字节(B)的存储空间,换算为360 KB,因为两者本质都是文本文件,所以大小一样。

3.5 随机1000条PE150测序reads的大小(fastq.gz)
占用了计算机53207、54992字节(B)的存储空间,换算为52、54KB,此时两者的大小略有差异,是由于压缩本身导致的,解压后恢复正常。
![]()
3.6 总结
0.3 M个碱基(bp),采用PE150测序,会分别产出1000条R1方向、1000条R2方向的reads。此时以txt、fastq格式存放,会占用计算机360 KB的存储空间。压缩成fastq.gz后,R1、R2方向的reads分别占用计算机 52 KB、54 KB的存储空间。
Fastq.gz文件大小 = ~ Fastq文件大小 / 6.6,压缩比为6.6。
4、网上几个测序数据量计算题目
1、双端测序换算
PE150或2×150,即双端测序,每条read长度150bp。
150bp×2端 × read数 = 数据量
例如,测50M read,150bp X 2端 X 50M read = 15000M = 15G(这个G表示的是碱基个数,而不是计算机的储存单位GB)。
2、单端测序换算
SE50或1×50,即单端测序,每条read长度50bp。
50bp ×1端 × read数 = 数据量
例如,测20M read,50bp ×1端 × 20M read = 1000M = 1G
3、计算多少条reads
问题描述:RNA-Seq数据,101bp大小。测序公司给我1.5G数据,大约有多少reads?
答:按照PE测序101和1.5Gbp计算,就是1.510^9
/100=1.5 * 10^7(条reads)= 15M条
参考资料
1、人体30亿个碱基对的基因组,容量有多少兆?
2、有关测序的数据储存问题
3、fastq压缩之后的gzip文件大小与样本数据量