hive随机抽取100条数据_使用TCGA数据随机抽取基因形成虚拟panel计算TMB
为了完成这个小探索,遇到了一个以前从来没有注意的问题,就是不同数据库对基因注释的记录差异问题。
https://mp.weixin.qq.com/s/2QJvQxVECcxpJIsId1pHYA
https://mp.weixin.qq.com/s/MDpX3tWQ7dojy84WjGuoZg
前些天朋友圈被刷屏的一个研究(https://mp.weixin.qq.com/s/aKt0RRNFo9NydLJbAQbTjw),提到了利用外显子组测序计算TMB是“金标准”,然而临床难以常规应用。基于二代测序技术基因组合(NGS panel)估测TMB是可行的替代手段,但如何选择临床适用的NGS panel尚缺乏有效的研究证据。
所以研究者基于TCGA数据库,研究随机抽取10~700个基因形成虚拟NGS panel,结果显示,当包含的基因数≥150个时,NGS panel计算的TMB与WES计算的TMB有较好的相关性;纳入同义突变可使相关性进一步提高。
这样就最好确立了包含150基因的NGS panel来做临床TMB检验, 如下图
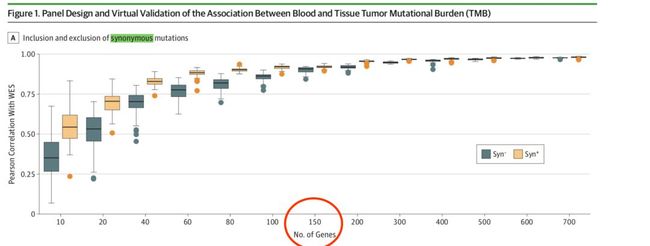
那我们来重复一下这个随机抽取10~700个基因形成虚拟NGS panel来计算TMB的过程吧。
下载TCGA的某种癌症的maf文件
maf格式文件我就不过多解释了,里面存放着该癌症的所有样本的所有的somatic突变位点。
比如乳腺癌的somatic mutation (SNPs and small INDELs)
MuSE Variant Aggregation and Masking (n=983)
MuTect2 Variant Aggregation and Masking (n=986)
SomaticSniper Variant Aggregation and Masking (n=975)
VarScan2 Variant Aggregation and Masking (n=986) GDC Hub
这里就选择GATK团队的MuTect2软件结果吧,下面的下载链接是有规律的url,记住,有规律哦。
https://gdc.xenahubs.net/download/TCGA-BRCA/Xena_Matrices/TCGA-BRCA.mutect2_snv.tsv.gz
探索maf文件
首先使用简单的shell命令来检查第9列:
cut -f 9 TCGA-BRCA.mutect2_snv.tsv |tr ';' '\n' |sort |uniq -c|sort -k1,1nr
当然,也可以使用大家熟练的R语言,反正后续绘图可视化还是R语言。
62288 missense_variant
22858 synonymous_variant
8869 3_prime_UTR_variant
7118 intron_variant
5734 stop_gained
5078 frameshift_variant
3471 splice_region_variant
2932 5_prime_UTR_variant
2206 non_coding_transcript_variant
1954 non_coding_transcript_exon_variant
1030 splice_acceptor_variant
870 downstream_gene_variant
776 splice_donor_variant
725 upstream_gene_variant
582 inframe_deletion
360 inframe_insertion
318 NMD_transcript_variant
249 coding_sequence_variant
177 protein_altering_variant
93 stop_lost
88 start_lost
72 stop_retained_variant
22 mature_miRNA_variant
5 intergenic_variant
4 regulatory_region_variant
1 effect
1 incomplete_terminal_codon_variant
可以看到,基因间区的突变位点其实都过滤了,所以这些突变位点都是要算在TMB里面。
这个时候有个很关键的问题,就是TMB里面的外显子长度定义问题,这里就选取参考文献的 38Mb吧:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5395719/
https://www.nature.com/articles/nm.4333/figures/5
得到基因的外显子长度之和
这里我GitHub项目:https://github.com/jmzeng1314/scRNA_smart_seq2/blob/master/RNA-seq/step7-counts2rpkm.R 里面探索过3种方法获取基因长度,然后发现 同样的基因在不同数据库记录的位置信息差距好离谱 所以不得不弃用 TxDb.Hsapiens.UCSC.hg38.knownGene 包。
这里还是使用CCDS记录文件吧,在数据库:ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/
wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.20180614.txt
cat CCDS.20180614.txt |perl -alne '{/\[(.*?)\]/;next unless $1;$gene=$F[2];$exons=$1;$exons=~s/\s//g;$exons=~s/-/\t/g;print "$F[0]\t$_\t$gene" foreach split/,/,$exons;}'|sort -u |bedtools sort -i >exon_probe.hg38.gene.bed
cat exon_probe.hg38.gene.bed|perl -alne '{$s+=$F[2]-$F[1]}END{print $s}'
## 计算得到 WES 全长是 36540331, 约 38Mb,所以就采用这个吧
cat exon_probe.hg38.gene.bed|perl -alne '{$s{$F[3]}+=$F[2]-$F[1]}END{print "$_\t$s{$_}" foreach keys %s}' > gene_length.human.txt
可以看到比较长和比较短的基因是:
$sort -k2,2nr gene_length.human.txt |head
TTN 114068
DST 44018
MUC16 43440
SYNE1 37090
MACF1 33953
RNF213 31550
HYDIN 31327
OBSCN 27932
DNAH8 27416
KMT2C 26711
jianmingzeng 17:53:15 ~/annotation/CCDS/human
$sort -k2,2nr gene_length.human.txt |tail
MTRNR2L4 86
RPL41 75
MTRNR2L10 74
随机抽取基因计算TMB
这个稍微有点难度,研究者随机抽取10~700个基因形成虚拟NGS panel 继续技术TMB,我们先看看原始的BRCA里面的TMB分布如下
rm(list = ls())
options(stringsAsFactors = F)
## UCSC xena source
BRCA.mutect2 = read.table( 'TCGA-BRCA.mutect2_snv.tsv.gz',sep = '\t',header = T)
colnames(BRCA.mutect2)
read.table()
head(BRCA.mutect2)
# 我们这里并没有区分同义突变和非同义突变
BRCA.mutect2$pos=paste0(BRCA.mutect2$chrom,':',
BRCA.mutect2$start,'-',
BRCA.mutect2$end)
TMB=as.numeric(table(BRCA.mutect2$Sample_ID)/38)
dat=data.frame(TMB=log2(TMB+1),BRCA='BRCA')
fivenum(dat$TMB)
library(ggpubr)
ggviolin(dat,x = 'BRCA', y = 'TMB',ylab = 'log2(TMB+1)',xlab='TCGA')
如下:
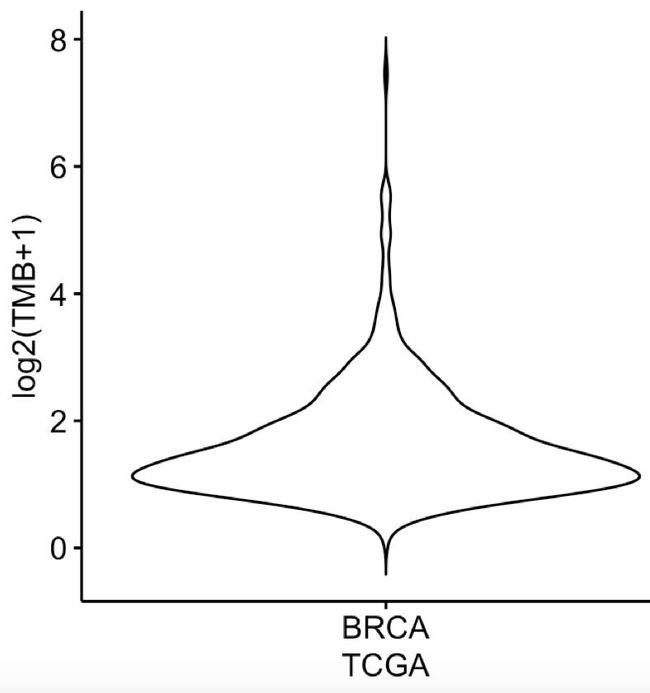
这个图在非常多的TMB文章其实都被分析过,只不过是它是跟TCGA数据库的所有其它癌症的TMB分布图画在一起的。
现在我们开始抽样,我们选择10-300个基因,每10个基因增加这样的panel进行随机抽样算TMB:
gl=read.table('gene_length.human.txt')
head(gl)
TMB=as.data.frame(table(BRCA.mutect2$Sample_ID)/38)
head(TMB)
allgenes=unique(BRCA.mutect2$gene)
tmp=lapply(seq(10,300,by=10), function(size){
as.numeric(lapply(1:1000, function(x){
cg=sample(allgenes,size)
panle_length=sum(gl[gl[,1] %in% cg,2])/1000000
small.BRCA.mutect2=BRCA.mutect2[BRCA.mutect2$gene %in% cg,]
small.TMB=as.data.frame(table(small.BRCA.mutect2$Sample_ID)/panle_length)
comp=merge(small.TMB, TMB,by='Var1')
# 第一个问题,这里忽略了很多TMB为0的样本,是否合理
cor(comp[,2],comp[,3])
# 第二个问题,计算TMB相关性的时候,我们并没有进行归一化。
}))
})
dat=do.call(rbind,tmp)
rownames(dat)=seq(10,300,by=10)
apply(dat, 1, mean )
dat=t(dat)
plot(apply(dat, 2, mean ))
library(reshape2)
df=melt(dat)[,2:3]
colnames(df)=c('number_of_genes','cor')
library(ggpubr)
ggviolin(df,x = 'number_of_genes', y = 'cor',
add = "boxplot")
出图如下:
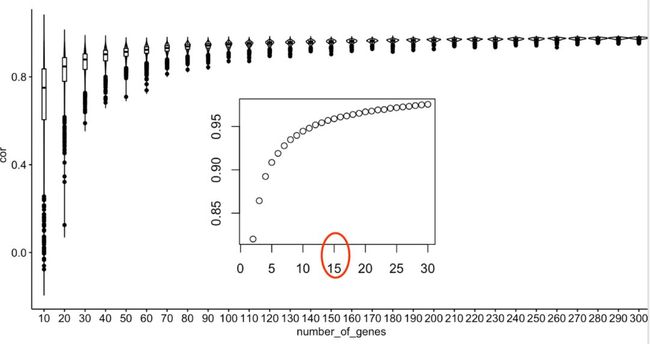
可以看到基因数量非常少的时候,panel得到的TMB跟WES得到的真实TMB的相关性波动性很大,而且差距很大,基因panel增加到150的时候比较稳定,而且相关性挺好的。
有3个小问题:
我们这里并没有区分同义突变和非同义突变
然后我们抽样的小基因panel会出现TMB为0的样本,我们并没有考虑它。
最后,计算TMB相关性的时候,我们并没有进行归一化。
有趣的是,结论差不多。
补充作业
作者也顺便算了一下TCGA数据库的所有其它癌症的TMB在150基因的虚拟panel与WES相关性分布情况,大家可以按照我前面在BRCA里面的方法完成下面这个图。

如果你愿意在我的指导下,完成这个小任务,那么请发邮件给我咯。
后记
TCGA教程我写不少了,但是本文的技术难度不足以位列28篇教程,作为一个小插曲发布。其它长期更新列表:
TCGA的28篇教程-使用R语言的cgdsr包获取TCGA数据(cBioPortal)
TCGA的28篇教程-使用R语言的RTCGA包获取TCGA数据 (离线打包版本)
TCGA的28篇教程-使用R语言的RTCGAToolbox包获取TCGA数据 (FireBrowse portal)
TCGA的28篇教程-批量下载TCGA所有数据 ( UCSC的 XENA)
TCGA的28篇教程-数据下载就到此为止吧
TCGA的28篇教程-指定癌症查看感兴趣基因的表达量
TCGA的28篇教程-对TCGA数据库的任意癌症中任意基因做生存分析
TCGA的28篇教程-整理GDC下载的xml格式的临床资料
TCGA的28篇教程-风险因子关联图-一个价值1000但是迟到的答案
TCGA的28篇教程-数据挖掘三板斧之ceRNA
TCGA的28篇教程-所有癌症的突变全景图
TCGA的28篇教程-早期泛癌研究
TCGA的28篇教程-CNV全攻略
TCGA的28篇教程-GTEx数据库-TCGA数据挖掘的好帮手
TCGA的28篇教程-所以癌症的突变全景图
另外,欢迎关注和推荐我们的全国巡讲给有兴趣的朋友
往期精选时间:3.16-3.18
生信技能树(爆款入门培训课)全国巡讲约你
生信技能树(爆款入门培训课)巡讲第一站-重庆 (已结束)
生物信息学全国巡讲之粤港澳大湾区专场 (正在报名)
生信技能树(爆款入门培训课)巡讲第二站-济南 (正在报名)
下一站是北京,预计3月30,31,以及4月1号这三天授课。