autoCell:用于scRNA-seq的分析工具

目录
- 简介
- 引言
-
- 相关工作
- 贡献
- 数据集和对比方法
- autoCell
-
- 高斯混合模型
- 图嵌入
- 结果
-
- 插补
- 去噪后轨迹分析
- 潜在特征捕获细胞病理学
- 发现细胞类型特异性分子网络
简介
动机:scRNA-seq使研究人员能够以单细胞分辨率研究基因表达。然而,由dropout引起的噪声可能会妨碍精确的数据分析。迫切需要开发可扩展的去噪方法来处理庞大但稀疏的scRNA-seq数据。方法:在这里,作者提出了autoCell:一种用于scRNA-seq插补和特征提取的高斯混合变分自动编码器网络。autoCell为大规模scRNA-seq或snRNA-seq数据的端到端分析提供了深度学习工具箱,包括可视化、聚类、插补和疾病特异性基因网络识别。结果:作者在模拟数据集和生物学相关的scRNA-seq数据上验证autoCell。结果表明,autoCell的插补提高了现有工具(论文中的轨迹分析方法为slingshot)在识别人类植入前胚胎(human preimplantation embryos)的细胞发育轨迹方面的性能。作者以阿尔茨海默症为案例,鉴定了疾病相关星形胶质细胞(DAAs,disease-associated astrocytes),并重建了DAA-specific的分子网络和参与细胞间通讯的配体-受体相互作用。
引言
单细胞技术是一项革命性的突破,使我们能够研究组织中每个细胞、每个状态下的基因组、转录组和多组学系统。结合荧光标记(fluorescent labeling)和显微切割(micro-dissection)等技术,它还可以确定空间属性和细胞间通信。这些技术被广泛使用,引发了基础医学和转化医学(basic and translational medicine)的革命。
单细胞或单核RNA测序(sc/snRNA-seq)对于从异质细胞中鉴定生物学和疾病相关的细胞类型和亚群非常重要。不同细胞状态下的基因表达的低维表征也可以重建细胞发育轨迹。然而,单个细胞中的mRNA含量是很少的,这需要近百万倍的放大。尽管测量技术得到了极大的改进,但技术因素仍然会在scRNA-seq实验所产生的数据中产生相当大的噪声,最明显的就是极低的捕获率。极低的RNA捕获率导致无法检测到一些已经有表达的基因,即dropout事件。dropout事件导致的假零计数与真零计数之间存在本质差异。考虑到稀疏的表达量,传统的分析工具无法达到科学的严谨性。
相关工作
深度学习算法在高维数据处理(包括稀疏基因组数据)中表现出令人信服的性能:
- DeepImute:基于深度神经网络的插值算法DeepImute就是例子。它使用dropout层来学习数据中的模式,以实现精确的插补。
- DCA:另一种去噪模型是通过深度计数自动编码器(DCA,depth-counting autoencoder)网络对scRNA-seq数据集进行去噪。DCA使用ZINB噪声模型将数据的计数分布(count distribution)、过度分散(over-dispersion)和稀疏性(sparsity)考虑在内。
- scScope:是一种可扩展的基于深度学习的方法,可以从数百万个异质的单细胞转录组数据中准确快速地识别细胞类型组成。
- scVI:与DCA和scScope相比,scVI使用变分自动编码器(VAE)来降低scRNA-seq数据的维数。然而,scVI实现的标准VAE仅在潜在变量上使用单一各向同性的多变量高斯分布,这通常不适用于表示多类别数据,如包含多种类型细胞的scRNA-seq数据。
- scVAE:是另一种基于VAE的方法,它使用高斯混合模型(GMM)作为先验分布,并引入泊松分布或负二项分布(NB)来获得细胞的潜在表示。然而,scVAE将GMM的均值和方差作为随机变量,并用具有参数 β \beta β的神经网络近似,这使得模型优化具有挑战。
随着sc/snRNA-seq数据分析挑战的增加,现在对更快和可扩展的去噪算法的需求变得迫切。
随着图神经网络的快速发展,图自动编码器可以学习图拓扑的低维表示,并使用全局的graph视图训练节点关系。越来越多工作考虑在sc/snRNA-seq数据分析中使用图神经网络框架。单细胞图神经网络(scGNN)通过图神经网络建立细胞-细胞关系,并使用混合高斯模型来建模异质基因表达的模式。scGNN还集成了三个多模态自动编码器。然而,scGNN框架需要更多的计算资源。
贡献
在这项研究中,作者提出了一个深度学习框架,即autoCell,用于从scRNA-seq数据进行插补和特征提取。准确度指数(accuracy index)表明,autoCell在模拟数据集和具有不同程度人类疾病的生物学相关sc/snRNA-seq数据集中优于六种最先进的插补方法。因此,autoCell是一种可扩展和精确的scRNA-seq数据处理方法,优于其他scRNA-seq数据分析工具。
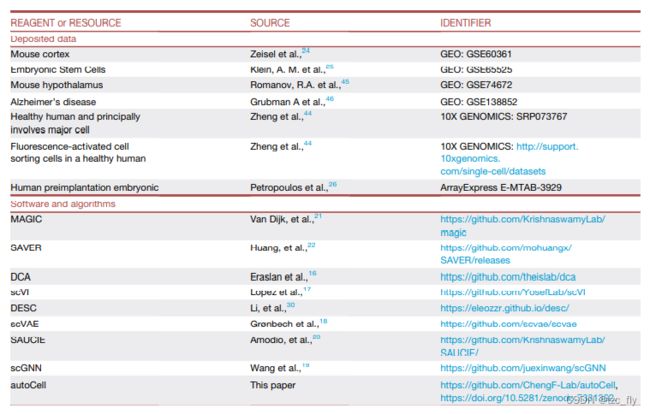
数据集和对比方法
数据集和对比方法见下表:
autoCell
autoCell的概述如图1所示。它是一个VAE网络,它结合了图嵌入和GMM来建模高维稀疏scRNA-seq数据的分布。通过集成GMM,autoCell可以更好地估计数据分布。作者应用图嵌入来处理sc/snRNA-seq数据。捕获局部数据结构的图信息是深度GMM的一个很好的补充,使网络成为具有局部结构约束的全局模型。最近的研究表明,ZINB分布是解决scRNA-seq数据dropout事件的合适工具。为了减少高度稀疏和过度分散计数数据中dropout事件的影响,作者引入了ZINB分布,从而对scRNA-seq数据进行去噪。
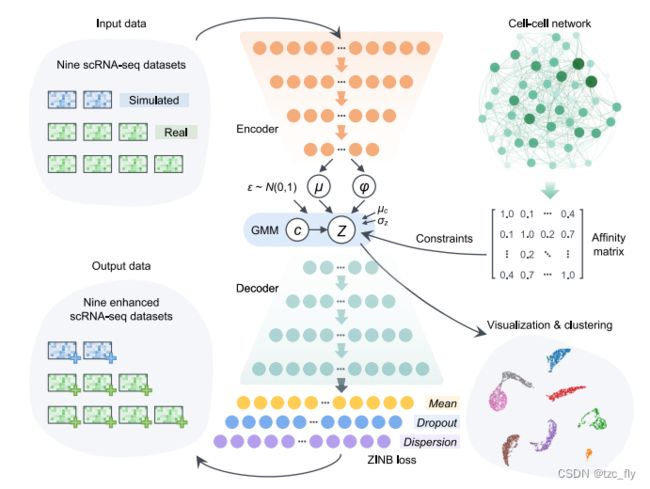
- 图1:autoCell使用高斯混合模型(GMM)和深度神经网络(DNN)来建模数据生成过程。它使用ZINB损失来处理scRNA序列中的dropout事件。编码器和解码器是一个两层神经网络,直接连接到输出的10维潜在变量(特征)。细胞-细胞网络用于约束潜在特征 Z;因此,相似的细胞具有相似的潜在特征和聚类分配。黄色节点表示负二项分布的平均值,这是表示去噪数据的主要输出,而紫色和蓝色节点表示ZINB分布的其他两个参数,即分散(方差)和dropout(零处的脉冲)。
高斯混合模型
使用高斯分布作为隐变量 z 的概率分布仅允许潜在表示一个模式(隐特征的每一维度只能属于一个高斯分布),虽然 z 是多元高斯分布,但这些高斯分布体现出单一的各向同性。GMM建模可以增加多样性。
对于一个 D D D维的训练样本集合 { X n } n = 1 N \left\{X_{n}\right\}^{N}_{n=1} {Xn}n=1N,我们聚类其为 K K K个簇。对于每个训练样本 x x x,我们学习一个隐特征 z ∈ R M × 1 z\in R^{M\times 1} z∈RM×1。我们假设特征遵循GMM分布。我们引入一个二值向量 c ∈ { 0 , 1 } K × 1 c\in\left\{0,1\right\}^{K\times 1} c∈{0,1}K×1指示隐特征属于的高斯分量。
ZINB分布在单细胞RNA数据分析中受到青睐。先前的研究表明,ZINB分布模型可以通过建模高度稀疏和过度分散的数据来有效地去噪单细胞RNA数据: Z I N B ( x ; λ , μ , ψ ) = λ I 0 ( x ) + ( 1 − λ ) N B ( x ; μ , ψ ) ZINB(x;\lambda,\mu,\psi)=\lambda I_{0}(x)+(1-\lambda)NB(x;\mu,\psi) ZINB(x;λ,μ,ψ)=λI0(x)+(1−λ)NB(x;μ,ψ)其中, λ \lambda λ代表零值的比例, μ , ψ \mu,\psi μ,ψ是NB的均值和方差。 I 0 I_{0} I0为0值得脉冲函数,当自变量为0时,值为1,否则为0。
生成模型:我们假设数据来自高斯混合分布。特别是,对于scRNA-seq数据 x x x,我们对生成过程建模如下: p ( c , π ) = ( p 1 , . . . , p K ) = ∏ k = 1 K π k c k p(c,\pi)=(p_{1},...,p_{K})=\prod_{k=1}^{K}\pi_{k}^{c_{k}} p(c,π)=(p1,...,pK)=k=1∏Kπkck p ( z ∣ c k = 1 ) = N ( μ k , d i a g ( σ k 2 ) ) p(z|c_{k}=1)=N(\mu_{k},diag(\sigma_{k}^{2})) p(z∣ck=1)=N(μk,diag(σk2)) P θ ( x ∣ z ) = Z I N B ( x ; λ , μ , ψ ) = λ I 0 ( x ) + ( 1 − λ ) N B ( x ; μ , ψ ) P_{\theta}(x|z)=ZINB(x;\lambda,\mu,\psi)=\lambda I_{0}(x)+(1-\lambda)NB(x;\mu,\psi) Pθ(x∣z)=ZINB(x;λ,μ,ψ)=λI0(x)+(1−λ)NB(x;μ,ψ)其中, c k , π k c_{k},\pi_{k} ck,πk为向量 c c c和 π \pi π的第 k k k个条目。 π k \pi_{k} πk遵循 ∑ k = 1 K π k = 1 \sum_{k=1}^{K}\pi_{k}=1 ∑k=1Kπk=1。 μ k \mu_{k} μk和 σ k 2 \sigma_{k}^{2} σk2代表第 k k k个高斯分量的均值和方差。 ( λ , μ , ψ ) = g ( z n ; θ ) (\lambda,\mu,\psi)=g(z_{n};\theta) (λ,μ,ψ)=g(zn;θ), g g g是一个参数为 θ \theta θ的神经网络。
推断模型:直接求解上述生成模型来找到隐变量的最大后验和参数的似然是困难的。我们使用新分布 q ϕ ( z , c ∣ x ) q_{\phi}(z,c|x) qϕ(z,c∣x)去近似后验分布 p θ ( z , c ∣ x ) p_{\theta}(z,c|x) pθ(z,c∣x)。该新分布可以用参数为 ϕ \phi ϕ的神经网络近似,我们假设 q ϕ ( z , c ∣ x ) q_{\phi}(z,c|x) qϕ(z,c∣x)可以分解为 q ϕ ( z , c ∣ x ) = q ϕ ( z ∣ x ) q ϕ ( c ∣ x ) q_{\phi}(z,c|x)=q_{\phi}(z|x)q_{\phi}(c|x) qϕ(z,c∣x)=qϕ(z∣x)qϕ(c∣x),因此有: q ϕ ( z ∣ x ) = N ( μ ~ , d i a g ( σ ~ 2 ) ) q_{\phi}(z|x)=N(\widetilde{\mu},diag(\widetilde{\sigma}^{2})) qϕ(z∣x)=N(μ ,diag(σ 2)) q ϕ ( c ∣ x ) = M u l t i n o m i a l ( π ~ ) q_{\phi}(c|x)=Multinomial(\widetilde{\pi}) qϕ(c∣x)=Multinomial(π )其中, [ μ ~ , d i a g ( σ ~ 2 ) ] = f ( x ; ϕ ) [\widetilde{\mu},diag(\widetilde{\sigma}^{2})]=f(x;\phi) [μ ,diag(σ 2)]=f(x;ϕ), π ~ = f ( z ; ϕ ) \widetilde{\pi}=f(z;\phi) π =f(z;ϕ), f f f为神经网络,参数为 ϕ \phi ϕ。
在生成模型和推断模型的框架中,参数可以被最大似然估计: m a x ϕ , θ ∑ i = 1 N l o g [ p θ ( x i ) ] max_{\phi,\theta}\sum_{i=1}^{N}log[p_{\theta}(x_{i})] maxϕ,θi=1∑Nlog[pθ(xi)]模型由 g g g和 f f f构成VAE。
图嵌入
图嵌入的目的是在样本相似图中找到低维特征,从而保持顶点对之间的相似性。通常,训练样本 { x n } \left\{x_{n}\right\} {xn}被视为相似图中的一个节点,相似图由亲和矩阵 W W W表示。优化后的特征 { z n ∗ } \left\{z_{n}^{*}\right\} {zn∗}为: { z n ∗ } = a r g m i n Z Z T = I ∑ i = 1 N ∑ j = 1 N w i j ∣ ∣ z i − z j ∣ ∣ 2 2 \left\{z_{n}^{*}\right\}=argmin_{ZZ^{T}=I}\sum_{i=1}^{N}\sum_{j=1}^{N}w_{ij}||z_{i}-z_{j}||_{2}^{2} {zn∗}=argminZZT=Ii=1∑Nj=1∑Nwij∣∣zi−zj∣∣22其中, Z = [ z 1 , . . . , z n ] Z=[z_{1},...,z_{n}] Z=[z1,...,zn], w i j w_{ij} wij代表亲和矩阵中的 ( i , j ) (i,j) (i,j)的值( ∑ j w i j = 1 \sum_{j}w_{ij}=1 ∑jwij=1)。如果样本在图上连接,那么它们的特征将彼此接近。因此,我们假设如果两个样本在图上连接,那么它们应该具有相似的潜在特征和聚类分配。因此,考虑图嵌入约束的最大似然为: m a x ϕ , θ ∑ i = 1 N ( l o g [ p θ ( x i ) ] − ∑ j = 1 N w i j d ( q ϕ ( z , c ∣ x i ) , q ϕ ( z , c ∣ x j ) ) ) max_{\phi,\theta}\sum_{i=1}^{N}(log[p_{\theta}(x_{i})]-\sum_{j=1}^{N}w_{ij}d(q_{\phi}(z,c|x_{i}),q_{\phi}(z,c|x_{j}))) maxϕ,θi=1∑N(log[pθ(xi)]−j=1∑Nwijd(qϕ(z,c∣xi),qϕ(z,c∣xj)))其中 d d d是衡量两分布距离的函数。
构造亲和矩阵:在近似图的嵌入方法中,亲和矩阵是重要的。亲和矩阵的一个典型选项是给定数据点的一组最近邻居,使用预定义的核函数来计算它们的相似性。例如,对于高斯核,亲和矩阵的元素定义如下:
如果 x j ∈ N ( x i ) x_{j}\in N(x_{i}) xj∈N(xi), N N N代表当前节点最近的 N s N_{s} Ns个邻居节点集合。 w i j = e x p ( − ∣ ∣ x i − x j ∣ ∣ 2 2 2 s i 2 ) w_{ij}=exp(-\frac{||x_{i}-x_{j}||_{2}^{2}}{2s_{i}^{2}}) wij=exp(−2si2∣∣xi−xj∣∣22)其中, s i s_{i} si是一个预定义标量。
结果
插补
在评估autoCell在估算缺失值方面的性能时,作者还选择了两个模拟数据和两个真实世界的 sc/snRNA-seq数据集作为基准,这些数据集具有良好注释的细胞类型。与几种最先进的算法相比,autoCell在模拟数据集和在真实数据集10%的人工丢失率下实现了由中值L1距离、余弦相似度和RMSE评估的较好的性能。此外,根据估计值和真实值的密度图,autoCell插补更接近真实表达值。总的来说,autoCell在sc/snRNA-seq数据插补分析中优于最先进的方法。
去噪后轨迹分析
除了识别细胞类型外,scRNA-seq还有助于按时间进程或发育阶段(即细胞轨迹)组织细胞。细胞从一种功能状态转变为另一种功能状态是发育过程中的关键。尽管目前存在一些模型可以根据scRNA-seq数据推断细胞发育轨迹,但大多数推断方法都没有解决dropout事件。作者测试了通过autoCell插值后推断scRNA-seq数据的细胞轨迹的准确性。使用了一个包含1,529个单细胞的基准数据集,这些单细胞具有从胚胎发生E3到E7的人类植入前胚胎发育的五个阶段的良好注释。在各种插值过程后使用slingshot重建了细胞发育轨迹。autoCell的插值在推断的伪时间和实时细胞发育之间产生了最高的对应关系。因此,autoCell在不同发育阶段捕获更准确的转录组动力学和细胞发育轨迹。
潜在特征捕获细胞病理学
作者还评估了autoCell推断的潜在空间能力,这在很大程度上反映了细胞之间的生物变异性,这些变异是基于先前通过无监督聚类将细胞分层为生物学上重要的亚群,然后进行人工检查和注释。作者将autoCell应用于两个模拟数据集和四个生物学相关的scRNA-seq数据集。这六个数据集的零比例在60%到90%之间。默认情况下,autoCell从输入数据中提取10个特征。为了公平比较,作者进一步应用了常见的scRNA-seq数据降维方法,包括scVI、DESC、scVAE、DCA 和SAUCIE,将输入数据降维到10维,并使用UMAP可视化从这些工具和原始数据中提取的特征。对于Klein数据集,scVI、scVAE和autoCell表现出更好的性能,DCA导致细胞类型d0和d2紧密相连。然而,SAUCIE和DESC只分离了细胞类型为d0的细胞,并错误地将细胞类型d7分为两种细胞类型。对于Zeisel数据集,作者发现autoCell、scVI和scVAE仍然优于其他模型。
作者将K-means聚类应用于autoCell提取的潜在特征,并通过与scVI、DESC、scVAE、DCA和SAUCIE进行比较来评估聚类精度。实验表明autoCell在所有测试的scRNA-seq数据集上显示出最佳性能。在Klein数据集中,使用autoCell的聚类输出与预定义的细胞类型注释(NMI=0.882,ARI=0.907)比排名第二的模型scVI(NMI=0.832,ARI=0.784)更为一致。在Zeisel数据集中,autoCell的聚类性能明显优于其他现有工具。总体而言,autoCell在捕获细胞病理生物学方面的准确度高于模拟和真实世界生物相关scRNA-seq数据集的现有最先进方法。
发现细胞类型特异性分子网络
在测试autoCell推断的细胞类型是否能够捕获人类疾病的特定病理生物学时,作者使用阿尔茨海默病(AD)作为原型,分析了星形胶质细胞(astrocytes)、小胶质细胞(microglia)、神经元和少突胶质细胞祖细胞(OPC)。总之,作者重新分析了从AD大脑和健康对照的内嗅皮层产生的13214个高质量细胞核。使用autoCell,作者确定了四个小胶质细胞簇、九个星形胶质细胞簇和五个OPC簇(图2A)。最近使用人类死后脑组织的研究确定了疾病相关星形胶质细胞(DAA)在AD发病机制和疾病进展中的关键作用。使用11个经实验验证的DAA标记基因(4个上调的标记基因[GFAP、CD44、HSPB1和TNS]和7个下调的标记基因[SLC1A2、SLC1A3、GLUL、NRXN1、CADM2、PTN和GPC5]),作者通过autoCell将星形胶质细胞亚群4鉴定为DAA。接下来,作者在人类蛋白质-蛋白质相互作用(PPI)网络模型下,使用最先进的基于网络的算法GPSnet构建了DAA特异性分子网络。DAA特异性模块网络包括由44种蛋白质连接的50个PPI,如APOE、MAPT、CD44、FOS和STAT3(图2B)。APOE和MAPT(微管相关蛋白Tau)是AD的两个最著名的风险基因。CD44是一种炎症相关蛋白。CD44的抑制可能是AD治疗的潜在策略。在一项小鼠模型研究中,Stat3缺失的星形胶质细胞表现出β-淀粉样蛋白和促炎细胞因子活性水平下降。DAA特异性分子网络中的蛋白质由多种AD相关途径富集,如细胞因子信号传导、脊髓损伤和脑源性神经营养因子信号传导途径(图2B)。例如,DAA特异性网络中的几种蛋白质(STAT3、MAPT、HSPB8、HSPB1、JUNB和LINGO1)富含多种细胞因子信号通路,包括IL-5、IL-2、IL-18、IL-3和IL-4,这与小胶质细胞介导的神经炎症在AD中的重要作用一致。因此,使用autoCell,作者可以识别与疾病相关的、细胞类型特异性的分子网络,这些分子网络参与了AD的关键病理生物学。
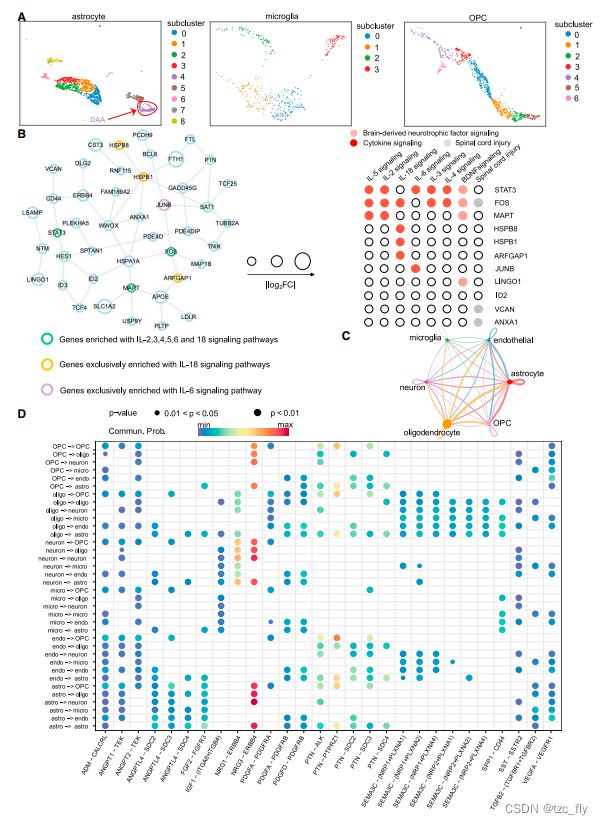
- 图2:使用autoCell发现阿尔茨海默病(AD)中细胞类型特异性分子网络和显著的配体-受体相互作用。
- A:星形胶质细胞astrocytes亚群(包括疾病相关星形胶质细胞DAA,即disease-associated astrocyte)、小胶质细胞microglia和OPCs的UMAP可视化,用autoCell簇标记。
- B:从人体蛋白质-蛋白质相互作用组重建DAA特异性分子子网络。DAA特异性模块网络包括由44种蛋白质(例如APOE、MAPT、CD44、FOS和STAT3)连接的50种蛋白质-蛋白质相互作用(PPI)。来自DAA特异性分子网络的蛋白质富含多种AD相关通路,包括细胞因子信号通路。
- C:使用CellChat推断细胞-细胞相互作用。
- D:在autoCell识别的细胞类型中,最重要的配体-受体对被选中。CellChat预测配体-受体相互作用。
作者还发现了AD中参与细胞-细胞通信的重要配体-受体相互作用。首先使用autoCell推断了细胞亚群,并使用CellChat预测了配体-受体的相互作用。如图2C所示,与其他三种细胞类型(神经元、小胶质细胞和内皮细胞)相比,作者发现星形胶质细胞、OPC和少突胶质细胞之间存在强烈的配体-受体相互作用。两个配体-受体对(NRG3-ERBB4和NRG1-ERBB4)显示了跨多个细胞-细胞对的强相互作用(图2D)。发现NRG3基因中的多个单核苷酸多态性与AD 的发病有关。此外,发现神经元中ERBB4的过度表达与AD神经病理学相关。最近的一项AD小鼠模型研究发现,NRG1和ERBB4的免疫反应性与海马区的斑块有关。使用AD作为典型例子,作者证明了autoCell识别的疾病相关细胞亚型可以识别参与AD发病机制的分子靶点和网络(即配体-受体相互作用),如果广泛应用,可以为AD或其他人类疾病提供潜在的药物靶点。