MDIPA:基于非负矩阵分解的MicroRNA-药物相互作用预测方法
MDIPA:基于非负矩阵分解的MicroRNA-药物相互作用预测方法
- 摘要
- 一、简介
- 二、方法
-
- 2.1 数据集
- 2.2 鉴定-药物相互作用
- 2.3 microRNA与药物的邻域信息
- 2.4 非负矩阵分解(NMF)
- 2.5 评价
-
- 2.5.1 使用独立数据集进行计算
- 2.5.2 评估与交叉验证
- 2.7 案例研究
- 2.8分子对接
-
- 2.8.1 结构的制备
- 2.8.2 分子对接研究
- 三、结果
-
- 3.1 MDIPA预测
- 3.2 使用独立数据集进行计算
- 3.3 参数分析
- 3.4 评估与交叉验证
- 3.5 案例研究
摘要
研究动机:有证据表明,microRNAs是一种小生物分子,它调节基因的表达水平,在疾病的发生和治疗中发挥着重要作用。药物作为重要的化合物,可以与microRNAs相互作用并改变其功能。对microRNAs-药物相互作用的实验鉴定既费时又昂贵。因此,开发有效的计算方法来预测microRNAs-药物相互作用是很有吸引力的。
结果:本研究提出了一种基于矩阵分解的microRNAs-药物相互作用预测方法(MDIPA),用于预测microRNAs与药物之间未知的相互作用。具体来说,MDIPA利用实验验证的药物与microRNA之间的相互作用、药物相似性和microRNA相似性来预测未被发现的相互作用。构建基于路径的microRNA相似矩阵,利用药物的结构信息建立药物相似矩阵。为了评估MDIPA的性能,在独立数据集和交叉验证下,将我们的MDIPA与四种最先进的预测方法进行了比较。两种评价方法的结果都证实了MDIPA的性能优于其他方法。最后,乳腺癌案例研究中的分子对接结果证实了我们的方法的有效性。综上所述,MDIPA可以有效预测潜在的microRNAs-药物相互作用。
一、简介
药物-靶标相互作用的实验研究既费时又昂贵。更糟糕的是,有时候,在投入大量资源之后,努力并不能发现新的交互作用。因此,在药物发现中,补充甚至替代的方法是值得的。药物重新定位和药物靶标预测是确定现有药物和药物化合物新应用的捷径。研究结果可为进一步的实验药物发现提供有效依据。
疾病是一种异常情况,有时与基因表达的变化密切相关。基因-疾病调控网络的研究表明,各种元素在这些网络中发挥着重要作用。例如,不同的化学物质,包括活性元素、辅助因子和药物,可以改变基因的表达水平。其他在基因表达网络中活跃的元素包括转录因子(TFs)、siRNAs(小抑制rna)和microRNAs (miRNAs)。例如,microRNAs与基因的调控元件或非翻译区域结合通过RNA干扰(RNAi),抑制mRNA翻译为蛋白质或诱导mRNA转录,进而改变基因表达水平。
由于药物在设计和作用模式上的多样性和灵活性,药物是影响基因表达和治疗疾病最常用的化学物质。microRNAs在基因表达和疾病中的作用是指通过影响microRNAs的功能来控制因microRNAs功能异常而引起的疾病。药物可以有效地达到这个目的。因此,识别药物和microRNAs之间的相互作用是很重要的。实验和计算方法都可用来识别这些相互作用。然而,由于实验方法需要时间和成本,在这些研究中的应用有限。另外,计算方法由于其灵活性和高吞吐量的结果而吸引人。
不同的计算方法已经被开发用来识别和预测药物- microrna相互作用。
一般来说,药物和microRNA的相互作用矩阵是非常稀疏的,其中未知的相互作用往往超过已知的相互作用。在各种交互预测算法中,基于相似度的方法应用最为广泛。他们的假设是,如果药物X与microRNA P相互作用,那么类似于药物X的药物也会与microRNA P相互作用,反之亦然。矩阵分解是将microRNA-药物相互作用矩阵Y分解成A和B两个矩阵,使用药物和microRNA的相似矩阵来预测未知的microRNA和药物的相互作用,其中Y≈A×B = Y*。矩阵因子分解可用于预测药物与靶点(如microRNA、蛋白质等)之间的相互作用。它通常是一种具有局部信息的最优全局方法。
所有讨论的方法都利用了药物靶标(drug-microRNA)直接关联。这很有挑战性,因为关于交互的已知信息是不完整的。针对现有方法的局限性,本研究旨在克服药物和microRNA关联预测方法的不足,加强矩阵分解方法,同时减少未知相互作用的负面影响。总的目标是识别microRNAs和药物之间新的相互作用,在这里microRNAs和药物的邻居信息被使用的最多。
二、方法
在这项工作中,鉴别药物和microRNA之间未被发现的相互作用被认为是一个基质完成的问题。基于构建的microRNA-药物相互作用矩阵,我们使用加权非负矩阵分解来预测药物和microRNA之间潜在的相互作用。在这种矩阵中,每个条目都是一个microRNA-药物相互作用的概率。基于药物和microRNA之间验证过的相互作用,我们构建了一个初步的相互作用矩阵(邻接矩阵)。然后,我们将这个矩阵扩展到新的microRNA和基于microRNA和药物相似矩阵的药物。

链接: NMF分解link.
链接: link.
为了评价MDIPA,将其结果与现有四种药物-靶标相互作用预测算法的结果进行比较。一个独立的数据集也被用来证实MDIPA可以比现有的方法更好地预测已知交互作用。此外,一个单独的案例研究的新预测通过分子对接方法得到验证。图1示意性地描述了MDIPA。

图1:MDIPA的示意图:
(a) MDIPA利用已知的microRNA-药物相互作用信息构建邻接矩阵Y。
(b)根据miRNAs与药物的邻居信息更新相互作用矩阵Y(见2.4节)。
©使用microRNAs与药物的相似矩阵,将矩阵Y分解为矩阵A和B (d),将交互作用矩阵Y扩展为包含新microRNAs与药物交互作用得分的预测矩阵Y* (e)。其中包含了新的microRNA和药物的交互得分。
(f) MDIPA提供交互
2.1 数据集
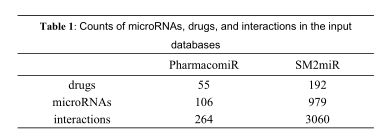
通过SM2miR 和PharmacomiR 检索药物与microRNAs的有效相互作用。
表1总结了每个数据集的统计信息。第一个数据集用于交叉验证评估,而第二个数据集用于独立测试数据集。此外,成熟microRNA和获批准药物的信息分别从miRBase和DrugBank 检索。microRNA-疾病关联信息从HMDD检索(http://www.cuilab.cn/hmdd)。
2.2 鉴定-药物相互作用
microRNA和药物的相互作用用一个矩阵表示,其中行(列)代表microrna(药物)。这种矩阵是基于实验验证的相互作用检索得到的从2.1节中描述的交叉验证数据集。
在这种矩阵中,如果microRNA i和药物j的相互作用被实验验证,一个元素(i,j)为1;否则,它是0。对应于这些0的相互作用被认为是未知的。
2.3 microRNA与药物的邻域信息
M = {m1, m2,…, mm} and D = {d1, d2,…,dn} 表示microRNA和药物的集合
MicroRNA-drug交互矩阵 (一个表示microRNA和药物之间的相互作用的邻接矩阵):
![]()
注意:如果microRNA mi与药物di 相互作用,则Yij=1;否则Yij=0。

在矩阵Y中,第i行(第j列)表示microRNA mi(药物di)的交互配置文件。
需要注意的是,microRNAs与无已知关联的药物之间的交互配置文件均为无,这可能导致无法预测药物与microRNAs之间潜在的相互作用。
为了克服这个问题, 对于 microRNA mz(药物dl),它与已知k个最接近(最相似)的microRNA(药物)交互配置文件的相似性,用于获得其初始相互作用矩阵,即:



2.4 非负矩阵分解(NMF)

传统的矩阵分解学习是基于欧几里得空间的,这就是为什么这种矩阵分解有时不能准确识别数据空间结构。为了提高学习性能,Xiao等人提出了图正则化非负矩阵分解。该方法基于图拉普拉斯正则化和Tikhonov项,后者保证了矩阵A和B的光滑性,图正则化主要是保证数据空间的判别结构。因此,非负矩阵可以重新表述为以下优化问题:

交互得分的计算从两个随机的非负(0到1)矩阵开始,即
![]()
求解优化问题(5)时,根据以下更新规则更新了这些矩阵:

收敛后得到预测矩阵为
![]()
其条目为MDIPA预测的每个microRNA-drug对的相互作用得分。
2.5 评价
为了评估我们提出的方法的性能,我们遵循两种策略:使用独立数据集评估和交叉验证(CV)。两种评估都需要两种类型的数据样本,阳性样本和阴性样本。本研究采用1481个有效的交互作用作为阳性样本。相互作用矩阵中的非相互作用对可以看作是负样本。然而,其中一些对可能是未知的,可能是相互作用的。为了筛选非相互作用的配对,我们使用了Luo等人描述的方法来排除阳性样本的最近邻居。然后我们随机选择负样本数据集数量相等的正样本的评价与一个独立的数据集和MDIPA与其他算法的比较包括CMF, NRLMF, GRMF 和NetLapRLS。
2.5.1 使用独立数据集进行计算
为了确保独立数据和CV数据之间没有重叠,我们使用BLAST在两个数据集中识别生物学上相似的microRNA。我们从这个数据库中删除了与CV数据集中相同的超过30%的microRNAs。最终,独立数据集由45对microRNA-drug对作为阳性样本和45个阴性样本组成。通过预测数据集内配对的交互得分来评估该方法的性能。已知阳性和阴性样本的交互作用得分分别为1和0。期望一个可靠的算法能够准确预测阳性样本的交互得分接近1,阴性样本的交互得分接近0。因此,算法的性能是通过预测阳性(阴性)样本的最大数量的最高(最低)交互分数的能力来评估的。
2.5.2 评估与交叉验证
用CV验证交互作用。方程1、2、4、5、6包含多种参数。我们利用CV通过网格搜索来设置这些参数。参数可能的值为:α,从{0,0.5,1}衰减率;k为{1,2,…,10}的邻域大小;r,矩阵A和B在{50,100,150}中的秩;λl从{0,0.25,0.5,1,2};λm和λd从{0,0.0001,0.001,0.01,0.1}。我们对这些参数的每个组合执行10倍的CV。然后,选择最佳组合。
我们使用10倍CV对MDIPA在三种不同场景下进行评估,如下:
CV1:microRNA和药物配对的相互作用的CV,每一轮随机选取Y中90%的阳性和阴性元素作为训练数据,剩余10%作为试验数据。
CV2:microRNA的CV,随机选择Y中90%的行作为训练数据,剩余的10%作为测试数据。
CV3:简历毒品,随机选择Y中90%的列作为训练数据,剩余的10%作为测试数据。
在每一轮中,我们将输入数据随机分为10个子集,其中10个中的一个用作测试数据集。将该子集中邻接矩阵得分为1的已知交互替换为0,以检验本文方法能否识别其交互得分。为了评估结果,我们在0.01的步骤中考虑从0到1的阈值。输入数据中有正样本和负样本两种情况。对于邻接矩阵中值为0的负样本,我们认为新值小于阈值的条目为真阴性,否则为假阳性。另一方面,对于邻接矩阵中被0代替的正样本,交互矩阵中的条目如果其值高于阈值则为真正,否则为假负。
基于这四种分类,我们计算评估指标,包括准确性、敏感性、特异性和精度。这些指标的所有使用的参数组合在我们的算法是计算。我们根据ROC曲线、ROC曲线下面积( AUC)和AUPR来选择最佳性能。
2.7 案例研究
MDIPA的能力是通过一个案例研究乳腺癌。MDIPA做出的预测互动得分接近1的人更有可能得乳腺癌。对60种潜在有趣的相互作用进行分子对接,以评估这些预测的相互作用得分。
2.8分子对接
分子对接技术预测化学化合物及其目标在生物系统中的原子水平行为,并计算结合自由能。对于乳腺癌案例研究中预测的硅验证,分子对接分为以下两个步骤进行。
2.8.1 结构的制备
乳腺癌病例研究中候选药物和microRNAs的结构需要在分子对接前进行优化。这些药物的初步3D结构从DrugBank检索。由于miRBase,RNAfold 和Vfold,获得候选microRNA的序列信息和初始三维结构。然后,用阿佛加德罗优化这些药物和microRNA的初步3D结构。利用经典的UFF力场和阶跃递进法进行了结构优化。然后在下一步中使用这些结构。
2.8.2 分子对接研究
为了证实MDIPA的结果,我们使用乳腺癌病例研究中涉及致癌microRNA的预测的相互作用药物- microRNA对进行分子对接分析。与生成的3D结构对接模型是使用AutoDockVina创建的。通过考虑最佳结合能,得到了相互作用模型。所有的准备过程和模拟对接都是通过AutoDockTools完成的。
三、结果
在本节中,我们将讨论基于独立数据集的MDIPA与其他现有方法的彻底比较。此外,本节还介绍了交叉验证的结果和一个案例研究,以确认MDIPA在鉴别乳腺癌背景下microRNA和药物的潜在相互作用方面的性能。
3.1 MDIPA预测
矩阵Y*包括1706个microRNA和2113种药物之间的所有相互作用得分。补充表S1列出了100种 microRNA-药物相互作用,MDIPA预测得分最高(图1,面板f)作为预测相互作用的例子。从表中可以看出,前50名中有43名,前75名中有64名,前100名中有87名得到了现有实验文献的证实。
3.2 使用独立数据集进行计算
使用独立数据集评估MDIPA的性能,并与2.5节中列出的最近的交互预测算法进行比较,以说明哪一种方法最能够预测独立数据集中microRNA-drug对的交互得分。如表2所示,MDIPA能够准确预测阳性(阴性)样本最高(最低)分的数量最多。补充表S2和S3包含了独立数据集中的所有正样本和负样本,它们的预测分数来自不同算法。
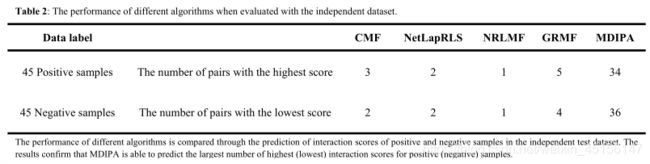
通过预测独立测试数据集中阳性和阴性样本的交互得分,比较不同算法的性能。结果证实,MDIPA能够预测最大数量的最高(最低)交互得分阳性(阴性)样本。
3.3 参数分析
正如第2.5节所讨论的,我们使用CV来设置各种参数。这些参数的值会影响收敛的运行时间。因此,每个参数的最优值至关重要。我们从初始值等于每个可能值范围中的最小值开始,然后增加这些参数的值。网格搜索的结果如下。虽然MDIPA在不同的k范围内产生的结果相对稳定,但当药物和microRNA的邻居数设置为7时,表现最好。
此外,我们注意到衰减速率阿尔法的增加可以提高预测精度。MDIPA展示了阿尔法为1时的最佳性能与价值。
对于
![]()
通过设置以下参数达到最好的性能:
![]()
此外,设置对于矩阵A和B的秩,设r=150在不同值之间表现最好。
3.4 评估与交叉验证
为了进一步比较MDIPA和2.5节中列出的算法的预测能力,我们使用了三种CV场景。这在之前提到过。交叉验证方案CV1旨在基于已知交互作用预测未知(drugi, microRNAj)对。相反,CV2和CV3设置的目标是识别没有已知相互作用的microRNA和药物。因此,CV1的任务比CV2和CV3的任务更容易。补充表S4总结了不同算法在三种不同CV设置下的AUPR和AUC值。从表中可以看出MDIPA算法相对于其他算法的优越性能。MDIPA获得了药物- microrna相互作用预测的AUC值为0.9586。尽管其他场景的AUC值较低,但MDIPA可以获得最高的值(0.8631和0.7614)。为了进一步显示MDIPA的性能改善,我们计算AUPR,并在补充表S4中显示。MDIPA在三种CV场景下分别得到最佳值0.9431、0.77574和0.7136。行(列)用于培训和测试场景CV2 (CV3)。由于正样本和负样本的数量可以行(列)变化,CV2和CV3的样本是不平衡的,因此AUPR值很低。
图2描述了CV1情景下不同预测方法的ROC曲线。补充图S1和S2展示了CV2和CV3两种场景下不同预测方法的ROC曲线。从图中给出的AUC值可以看出,MDIPA优于其他竞争方法。

3.5 案例研究
MDIPA的能力将通过一个案例做进一步研究,该案例涉及预测药物和乳腺癌中microRNA之间的相互作用。有许多抗癌药物如多烯紫杉醇,吉西他滨,阿霉素,紫杉醇,戈舍瑞林,和吉非替尼,有效地利用在乳腺癌的治疗中(图3),图3展示了乳腺癌致癌microRNA与其潜在相互作用药物之间的相互作用网络。。在我们的案例中,有18种致癌和32种抑制肿瘤的microRNA与乳腺癌相关。

图3中的一个具体例子是,预测化疗药物吉西他滨与mir-18a、mir-191、mir-93、mir-106a、mir-9、mir-182、mir-25、mir-22和mir-375的相互作用因子。补充图S3和S4说明了这两种相互作用的分子对接。紫杉醇是另一种化疗药物,被鉴定为可能与mir93、mir-18a和mir-221相互作用的分子。补充图S5和S6提供了其中两种相互作用的分子对接确认。补充表S7总结了用于分子对接的预测相互作用的结合自由能。图S7到S12进一步展示了致癌microRNA及其相互作用药物的分子对接例子。总的来说,候选药物- microRNA对的分子对接结果支持了MDIPA的预测性能。
所有数据与代码链接: link.