GAERF: predicting lncRNA-disease associations by graph auto-encoder and random forest(通过图自动编码器和随机森林)
GAERF: predicting lncRNA-disease associations by graph auto-encoder and random forest(通过图自动编码器和随机森林预测lncRNA-疾病的关联)是2021年才接收的论文,由安徽大学郑老师组的发表在Briefings in Bioinformatics上
文章目录
- 摘要
- 一、Introduction
- 二、Materials and methods
-
- 1.Matrix representation
- 2.Construction of LMD network
- 3.GAERF
- 三、Results
- 四、个人总结
摘要
预测疾病相关的长链非编码RNA(lncRNAs)有利于发现新的生物标志物,用于预防、诊断和治疗复杂的人类疾病。在本文中,我们提出了一种基于机器学习技术的分类方法,通过图自动编码器(GAE)和随机森林(GAERF)来识别疾病相关的lncRNA。首先,我们将lncRNA、miRNA和疾病的关系构建成一个异构网络。然后,GAE从网络中学习节点的低维表示向量,降低了生物数据的维数和异质性。将这些特征向量作为输入,我们训练了一个随机森林分类器来预测新的lncRNA-疾病关联。相关实验结果表明,该方法能够准确地表示lncRNA-疾病特征。由于集成学习方法,GAERF获得了优异的性能,显著优于其他方法。此外,案例研究进一步证明了GAERF是一种有效的预测lncRNA-疾病关联的方法。
一、Introduction
长非编码RNA(lncRNAs)由200多个核苷酸组成,长度没有明显的蛋白质编码潜力[1,2]。根据基因编码数据库的最新估计,人类基因组包含16 000个编码28 000多个不同基因的基因。然而,只有少数lncRNAs具有指定的功能[4]。
随着lncRNA研究的发展,越来越多的证据表明失调的lncRNA与多种疾病有关[5,6]。例如,据报道,MALAT1在多种癌症类型中过度表达[7,8]。MEG3 rs3087918与乳腺癌风险降低相关[9]。CRNDE通过促进POU2F1的表达促进肝细胞癌的进展[10]。SNHG5在鼻咽癌中作为肿瘤启动子[11]。SNHG7促进了鼻咽癌的增殖和迁移[12]。WWC2-AS1在辐射诱导的肠纤维化中高度表达[13]。LncRNA表达模式与急性髓系白血病的发展和进展密切相关[14]。这些研究表明,异常表达的lncRNAs可作为疾病诊断和预后的生物标志物以及癌症治疗的潜在靶点。
重要的是,越来越多的实验验证的关联使我们能够通过计算方法预测潜在的lncRNA关联。陈等人[15]首先提出了相似疾病往往与功能相似的非编码区带相关的假设,并发展了一种半监督方法,称为LRLSLDA。到目前为止,已经有大量的研究是基于数据库[16–20],研究人员提出了多种模型来预测LDAs[21–24]。这些模型中的一些基于随机游走预测疾病相关的lncrNA[25]。例如,陈等人[26]提出了一种称为IRWRLDA的模型,该模型基于改进的带重启的随机游走。IRWRLDA使用lncRNA表达相似度和疾病语义相似度作为随机游走的初始概率向量。于等人[27]开发了一个在有向双关系图上执行双随机游动的模型来预测LDAs。顾等[28]建立了预测LDAs的全局网络随机游走模型。这些模型可以应用于没有任何已知相关基因的疾病。
有些模型基于矩阵运算。傅等[29]构造了一种基于矩阵分解的方法,称为MFLDA。MFLDA通过矩阵三因子分解将异构数据源的数据矩阵分解为低秩矩阵,以探索和利用它们内在的共享结构。陆等[30]通过整合基因-疾病、基因-疾病和基因-基因关联,建立了诱导矩阵完成模型。宣等[31]提出了一个新的方法叫PMFILDA根据概率矩阵分解推断潜在的LDA。PMFILDA通过整合三个新构建的关联网络,构建了一个lncRNA-疾病加权关联网络,并基于疾病的语义相似度和lnc RNA函数的相似度,采用k近邻算法进一步更新。
另外,有些模型是基于深度学习的。宣等人提出了两个经典的基于CNN的预测模型,GCNLDA [32]和CNNDLP [33]。GCNLDA认为,一些模型未能深入整合包含lncRNAs、疾病和微小核糖核酸的异构网络[34–37]的拓扑信息,然后开发了图形卷积网络和卷积神经网络来学习lnc RNA-疾病对的网络和局部表示。CNNDLP认为,以前的大多数方法未能深入集成异构多源数据并从这些数据中学习低维特征表示,因此集成了来自异构源的多种数据,包括与lncRNAs、疾病和微小核糖核酸相关的关联、相互作用和相似性。与几种最先进的方法相比,这两种方法具有优越的性能。
虽然上述方法取得了很好的效果,但也存在一些局限性。基于随机游走和矩阵运算的方法很难挖掘出疾病网络中节点的拓扑信息;GCNLDA和CNNDLP模型的结构过于复杂,需要建立两个框架并调整许多参数,它们的性能可能会受到噪声(无关或低质量)数据的影响。为了解决这些问题,我们开发了一个基于图形自动编码器(GAE)和随机森林(射频)的LDAs预测模型(简称GAERF)。使用四个经典分类器和两种嵌入方法的大量对比实验证明了该方法的优越性能。此外,我们还利用外部数据集和实验验证的LDAs数据库来验证我们的模型的有效性。
简而言之,本研究有以下贡献:(1)利用已知的关联和前k个相似信息,创建一个由lncRNA、miRNA和疾病组成的异质网络(LMD网络),可以提供生物分子之间更精确的结构关系;(二)提出了一个结构简单的计算模型(GAE和RF)。GAE主要为图中的节点寻找合适的嵌入向量,并通过嵌入向量实现图的重构。得到的节点嵌入可以代表原始数据的主要成分,用于RF分类器;(3)提高计算模型的预测精度。
二、Materials and methods
在这项研究中,预测疾病相关lncRNA的数据集是从以前的工作中获得的[29,38]。数据集中有240个lncRNA、412种疾病和495个miRNAs,其中2697个LDAs来自Lnc2Cancer [39]、LncRNADisease [40]和GeneRIF [41]数据库,1002个lncRNA-MiRNA相互作用来自starBase [42]数据库,13562个MiRNA-疾病关联(MDAs)来自HMDD[43]数据库。
1.Matrix representation
给定l lncRNA、m miRNA和d疾病,它们的成对关联/相互作用分别由l×d LDAs矩阵LD、l×m lncRNA-miRNA相互作用矩阵LM和m×d MDAs矩阵MD表示。如果两种不同类型的生物分子之间存在关联/相互作用,则矩阵对应位置的元素值为1,否则为0。例如,如果ith lncRNA与jth疾病相关,则LD (i,j)=1,否则LD (i,j)=0
另外,lncRNA的功能相似矩阵按陈等[44]方法计算,∈R1×l表示;miRNA与疾病的相似矩阵按王等[45]的方法计算,分别用SML∈Rm×和SMD ∈ Rd×d表示。由于部分lncRNAs、miRNAs和疾病的相似度都为0,我们构建了基于LM和MD的高斯核相似度[23],并用高斯核相似度代替了SML、SMM和SMD中相应的0。最后,共有240×240个lncRNA相似性,495×495个miRNA相似性,412×412个疾病相似性。
2.Construction of LMD network
LncRNA和疾病的更多异质特征对于提高模型的准确性具有重要意义,但是一个没有相似性的模型是噪声数据,这将影响模型的性能。为了减少噪声数据,我们使用部分相似性得分来构建更精确的相似性矩阵。对于非负矩阵,向量SML (i),即矩阵SML的第I行,表示I本身和每个非负矩阵的相似性。我们只考虑前k个最相似的lncRNAs (i1,i2,…,ik) i n S M L (i) a n d认为它们将与lncRNA i相联系.同时,S M L (i)中的相应值,如SML(i,ij),j=1,2,…k设置为1,SML (i)中的其他值为0。经过预处理,我们得到了一个新的相似矩阵,表示为SL ∈ Rl×l,同样,我们得到了新的相似矩阵SM∈Rm×和SD ∈ Rd×dmiRNA和疾病。
基于关联矩阵和相似矩阵,我们可以构造LMD网络。LMD网络可以看作是一个无向图,它包括一个节点集和一个边集。每个节点代表一个基因,每个边代表连接的生物分子之间的关系。LMD网络的邻接矩阵可以表示为

其中LMT、L、DTT和MDT分别表示矩阵LM、LD和MD的转置。
3.GAERF

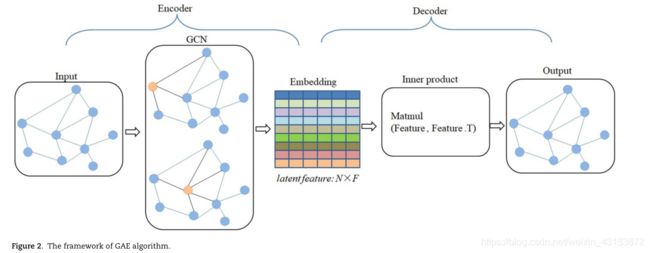
基于LMD网络,建立了图形嵌入和集成学习预测模型。GAERF的预测过程分为两个部分(如图1所示):GAE学习LMD网络的节点表示,RF预测潜在的LDAs。
图嵌入方法旨在从图中学习节点的低维表示向量[46–48]。有了这些表示向量,其他经典的机器学习算法可以应用于图形数据。GAE [49]在基于神经网络的图嵌入模型中,它使用GCN编码器和内积解码器来学习节点嵌入(见图2)。由于其简单的编解码结构和高效的编码能力,GAE被广泛应用于生物医学任务,如MDA预测[50,51],lnc RNA-miRNA相互作用预测[52],蛋白质-蛋白质相互作用预测[53]等。
给定LMD网络G的一个邻接矩阵H及其度矩阵d . 我们让 Z∈N×F表示 G中节点的随机潜在特征,其中N为节点数,F为特征维数。根据前人的工作[48],将F设置为100,将节点的原始特征初始化为单位矩阵I。
编码器是一个简单的两层图形卷积网络,定义如下:

其中∨H = D 1/2HD-1/2,ReLU(x)=max (0,x)指模型的激活函数,Wiis为第ith层的参数矩阵。
解码器计算两个节点之间的边的概率来重建图形

其中sigmoid(x)= 1 1+e外部参照激活函数,Z=GCN (I,H)。
为了测量重建图和原始图之间的差异,损失函数L被定义为

其中y表示邻接矩阵H中元素的值(0或1),n d y表示重构邻接矩阵H中相应元素的值(在0和1之间)。
通过GAE,我们获得了LMD网络中节点的潜在特征表示,每个lncRNA疾病对由一个200维向量表示。然后,实施RF来预测潜在的lncRNA-疾病关联。RF对噪声和稀疏数据具有较高的分类精度和良好的鲁棒性。它已被广泛应用于计算生物学。例如,陈等人[54]开发了一个用RF预测miRNA-疾病关联的模型。Dezs˝等人[55]提出了一种基于RF的方法来对蛋白质进行评分,以生成新靶标的可药物性评分。张等[56]利用RF从大规模,噪声和稀疏数据预测药物-药物相互作用。
RF是通过使用训练集构建大量决策树并输出结果来操作的(见图3)。它有三个重要参数,最大特征数、树数和最小样本叶大小。这些参数对RF的性能影响很大。然而,我们将树的数量设置为500,并将其他参数设置为默认值,GAERF可以实现出色的性能。经过训练,我们得到了一个可以推断的预测模型
通过对lncRNA-疾病配对进行评分来评估潜在的LDA。lncRNA-疾病对的较高分数表明lncRNA更有可能与疾病相关。
三、Results
老套路
四、个人总结
值的学习的点:
1、作者在整合相似性的时候,选取了前k个最大相似性值来用,来减少噪声,觉得这个处理数据的小方法还是不错的。
2、作者做的实验工作非常充足。
个人观点:
1、由于自身也是做lncRNA-disease关联预测的,看到这篇论文时,实在觉得没啥太大的创新,但是实验做的挺多,但也都是传统套路吧,有些是审稿人常要加的实验。简单来说,作者的方法包括两个部分,一部分使用图自动编码器提取lncRNA和疾病特征,第二部分用随机森林作为分类器来进行分类。
2、说一下和前人工作重复的点,(1)作者构建网络时,也是参考了GCNLDA的这篇文章的方法,以及BIB去年发的一篇VADLP(玄老师组常用的构建方法)的论文,包括使用的数据也是一样(都是从余老师(MFLDA)之前发的论文中提取来的)。(2)作者使用了图卷积相当于是采用GCNLDA这篇论文的一个分支。(GCNLDA包括两个分支,一个CNN,一个GCN)。(3)作者将GCNLDA和VADLP的工作中基于神经网络的分类器换成随机森林来分类。作者觉得GCNLDA和VADLP这两篇工作做的复杂,所以就简化了,相当于拿出人家的部分工作又重新做了一遍。感觉没有什么太大的创新。
仅为个人观点,并没有批判之意。