Chemical Science | Img2Mol+: accurate SMILES recognition from molecular graphical depictions

GitHub - bayer-science-for-a-better-life/Img2Mol
一、问题提出
Img2Mol能够正确地将高达88%的分子描述转化为他们的SMILES表示。大多数与药物挖掘相关的刊物以图片和表格的形式在文章中包含化学和生物测定数据,在新药物发现项目的开始阶段,研究人员面临着一个令人不快的事实,即最新的和相关的数据仍然作为原始数据隐藏在文章和专利中。
能够从图像中自动识别正确的分子内容仍然是一项非常具有挑战性的任务,但它可能对药物发现过程产生重大影响。成功的方法不仅必须捕获二维描述中包含的原子和键,而且必须将它们转换为有效的分子,理解原子类型并正确识别分子图。
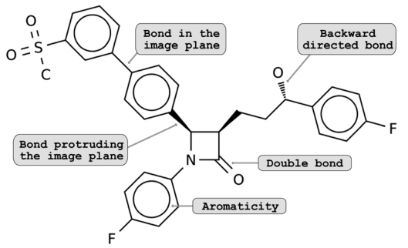
在某些情况下,原子的空间排列(即同分异构)由不同的键类型编码:位于panel上的键以规则的线表示;panel面下的键显示为虚线楔形,而从图像panel突出的键被放大或绘制为实心楔形。
二、模型
1、预训练
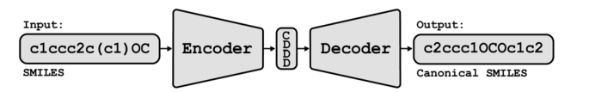
输入SMILES字符串被编码为512维特征向量(CDDD embedding)。decoder从CDDD embedding还原为规范的SMILES。
2、训练

损失为(l为均方误差):
![]()
3、数据集
1)Training set:ChEMBL25数据库和PubChem,使用RDKit规范SMILES表示并预处理。
筛选规则:删除了立体化学信息,删除了重复的分子,筛选有机分子(分子量在12到600 Da之间,超过3个重原子,logp在(-7,5)之间,处理过的数据集包含11 100 000个惟一的规范SMILES
训练集、验证集和测试集,验证集大小与测试集相同(50 000个惟一示例)
2)Benchmark datasets:
由于基线方法的计算时间较长,只使用了原始测试集的一半。该数据集由平均大小为25个原子的典型小分子组成,从6到44个原子不等。
STAKER:30 000张图像和分子描述。基于美国专利局(USPTO)的数据。图像分辨率是256*256 px。分子平均由24个原子组成,最小的是7个,最大的是51个
USPTO:基于美国专利局(USPTO)数据的4852张图像和分子描述的集合。649*417 px。由平均大小为28个原子的分子组成,从10到96个原子不等。
UoB:由伯明翰大学开发的5716张化学结构的图像和分子描述,从Rajan等获得。762*412 px。分子相当小,平均只有13个原子,从4到34个不等
CLEF:基于评估论坛(CLEF)的会议和实验室测试集,收集了711张图像和分子描述。1243*392 px。平均大小为26个原子的分子组成,从4到42个原子不等
JPO:日本专利局(JPO)数据收集的365张图片和分子描述。该数据集包含许多文本标签,包括日文字符,以及不规则特征,包括线粗细的变化。一些图像质量较差。图像的平均分辨率是607 * 373 px。分子平均由20个原子组成,最小的5个,最大的43个。
对于较小的基准数据集(USPTO, UoB, CLEF和JPO),通过添加旋转(随机从[-5,5])和剪切(x剪切因子随机[-0.1,0.1])
4、图像生成
图像由三个不同的化学信息学库生成:RDKit、OpenEye OEChem TK和Indigo,对于每个库,探索不同的描述设置:改变芳香性标记,添加原子编号,改变杂原子的键厚和字体大小,改变分子的方向,使用超原子(常见药物化学基团的文本缩写,如甲基的“Me”),或赋与键的表示风格等,输入分辨率随机选择在192像素到256像素之间随机。
MolVec,Imago and OSRA都是基于规则的。现有的基于深度学习的方法目前都无法进行比较。
三、实验Results
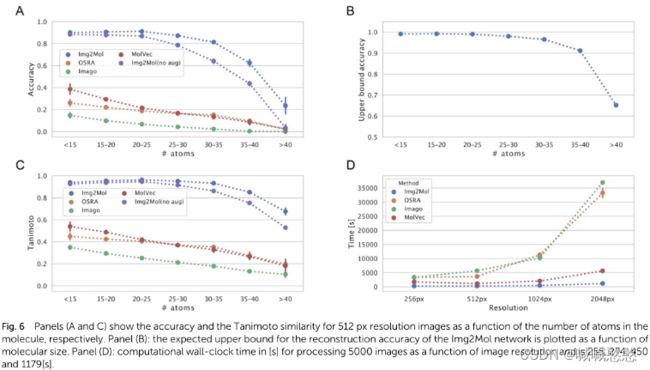
评价比较方法的性能:准确性和Tanimoto
A:更小、更简单的分子应该更容易提取。因为(i)分子图更容易处理(ii)对于恒定的输入图像大小,较小的分子具有更好的分辨率(大分子必须在相同的分配空间中包含更多的信息)。
B:对于非常大的分子,似乎可以预期的最大精度将在65%左右。假设Img2Mol总体性能的下降是由两个因素引起的:第一,分子识别任务变得更加困难,卷积网络产生的CDDD向量的均方误差随着分子的大小而增加。其次,CDDD解码器本身对于较大分子的性能较差。
D:显示在不同输入分辨率下四种方法的比较推理速度。
对于专利中的分子image,清晰度不高,特别是老专利:

健壮性的数据增强:从Img2Mol(no aug)到Img2Mol
Img2Mol成为唯一能够解码大约四分之一的大分子的方法(在这些情况下,Tanimoto的总体相似性为70%)
benchmark:

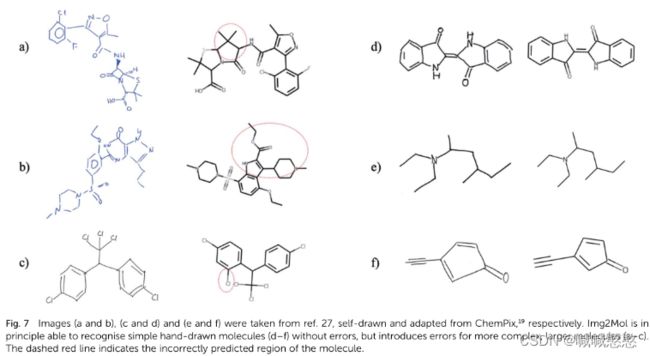
手写识别泛化:图def简单的分子结构可以泛化,但是复杂的就不能很好的泛化。