单细胞分析实录(4): doublet检测
最近Cell Systems杂志发表了一篇针对现有几种检测单细胞测序doublet的工具的评估文章,系统比较了常见的例如Scrublet、DoubletFinder等工具在检测准确性、计算效率等方面的优劣,以及比较了使用不同方法去除doublet后对下游DE分析、轨迹分析的影响。

现有的检测方法,基本都会先构造出虚拟doublet,然后将候选droplet与这些虚拟doublet比较,很相似的那些就定义为doublet。这里的虚拟doublet是通过随机组合两个(类)细胞的表达值得到的虚拟的doublet,可以作为检测时的参照。
在现有的9种方法中(Scrublet、doubletCells、cxds、bcds、Hybrid、DoubletDetection、DoubletFinder、Solo、DoubletDecon),文章的结论是DoubletFinder的准确率最高。
里面的方法我用过三种:Scrublet、DoubletFinder和DoubletDecon。前面两个用法类似,需要提供一个参数表示doublet的占比。DoubletDecon的原文我看过,算法比较简单,不需要提供doublet的占比,减少了用户额外的输入,不过也造成了一些麻烦,有时候会报告出很多doublet,多得惊人。实际分析中,我采用“两步走”的策略:选取了Scrublet和DoubletFinder的共同结果作为doublet去除掉,此外在后续聚类分亚群的过程中,根据一些已知的经典的细胞marker来判断doublet,比如CD45和EPCAM都高表达的亚群极有可能是doublet。
接下来,我会简单介绍DoubletDecon、DoubletFinder、Scrublet三个工具的使用。
1. DoubletDecon
该方法中间有一步用到了类似bulk RNA-seq里面deconvolution的思路来评估每一个细胞的表达模式,因此叫DoubletDecon。 这里用含有500个细胞的真实数据作为例子。在使用DoubletDecon之前,需要先用seurat对数据进行初聚类,seurat的使用我后面会详细讲,这里先把标准流程放上来。
library(tidyverse)
library(Seurat)
library(DoubletDecon)
test=read.table("test.count.txt",header = T,row.names = 1)
test.seu <- CreateSeuratObject(counts = test)
#Normalize
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
#FindVariableFeatures
test.seu <- FindVariableFeatures(test.seu, selection.method = "vst", nfeatures = 2000)
#Scale
test.seu <- ScaleData(test.seu, features = rownames(test.seu))
#PCA
test.seu <- RunPCA(test.seu, features = VariableFeatures(test.seu),npcs = 50)
#cluster
test.seu <- FindNeighbors(test.seu, dims = 1:20)
test.seu <- FindClusters(test.seu, resolution = 0.5)
test.seu <- RunUMAP(test.seu, dims = 1:20)
test.seu <- RunTSNE(test.seu, dims = 1:20)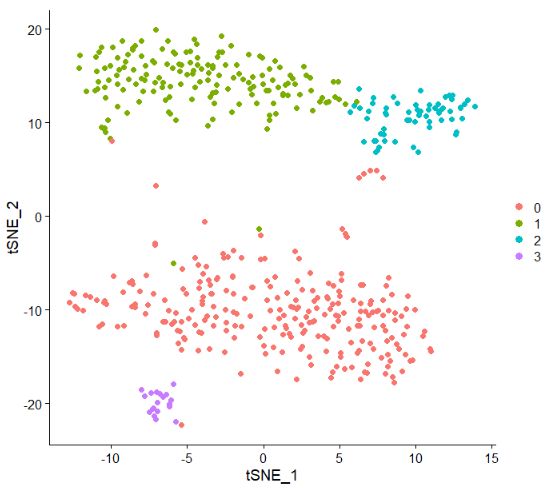
然后才是DoubletDecon的代码
#Improved_Seurat_Pre_Process()
newFiles=Improved_Seurat_Pre_Process(test.seu, num_genes=50, write_files=FALSE)这一步主要是找差异基因,会返回三个表格,分别表示marker基因的表达矩阵、所有基因的表达矩阵、细胞的seurat_cluster注释,前两个文件的第一行第一列有相应的注释。
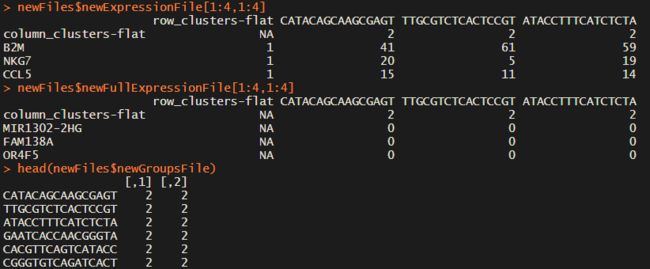
然后就是找doublet的主要步骤了
#Main_Doublet_Decon
results=Main_Doublet_Decon(rawDataFile=newFiles$newExpressionFile,
groupsFile=newFiles$newGroupsFile,
filename="tmp",
location="./",
species="hsa",
rhop=1,
num_doubs=80,
write=FALSE,
heatmap=TRUE,
centroids=TRUE,
nCores=2)这里面有几个很重要的参数,rhop默认值为1,用它来调节皮尔森相关系数的阈值(如下图)。在seurat聚类之后,这个软件会进一步将很相似的cluster合并,利用的就是最初cluster之间表达的相关性。这个值也很麻烦,前面提到的DoubletDecon会检测出很多doublet的问题可能就是这个参数设置不对导致的。那这个参数应该如何设置,可能软件作者也意识到了这个问题,后面又发了一篇Protocol,专门讲参数如何选择,核心思想就是多试几次。(事儿真多,准备放弃这个软件了)

接下来把DoubletDecon的检测结果保存成单独的文件,方便后面使用
doublet_df <- as.data.frame(results$Final_doublets_groups)
doublet_df$DoubletDecon="Doublet"
singlet_df <- as.data.frame(results$Final_nondoublets_groups)
singlet_df$DoubletDecon="Singlet"
DD_df <- rbind(doublet_df,singlet_df)
DD_df <- DD_df[colnames(test),]
DD_df$CB = colnames(test)
DD_df <- DD_df[,c("CB","DoubletDecon")]
write.table(DD_df, file = "DoubletDecon_result.txt", quote = FALSE, sep = '\t', row.names = FALSE, col.names = TRUE)也可以将doublet的结果投到tsne图上看看效果
[email protected]$CB=rownames([email protected])
[email protected]=inner_join([email protected],DD_df,by="CB")
rownames([email protected])[email protected]$CB
DimPlot(test.seu,reduction = "tsne",pt.size = 2,group.by = "DoubletDecon")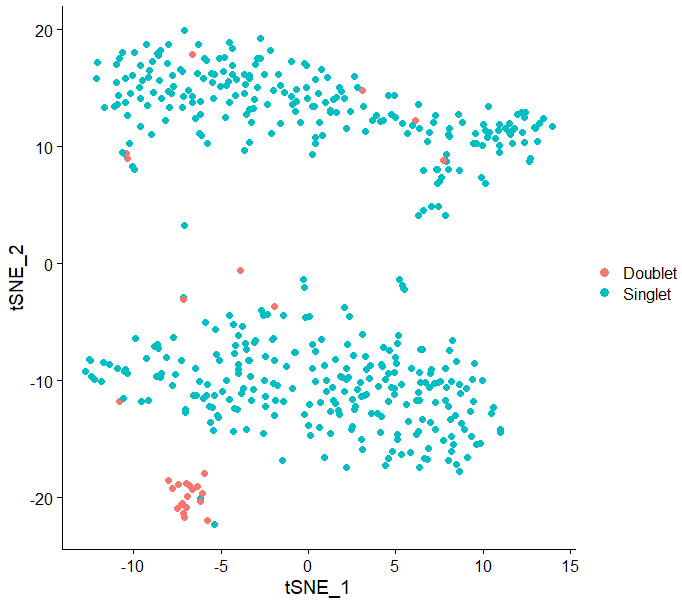
看上去还不戳,符合doublet单独成群的预期
2. DoubletFinder
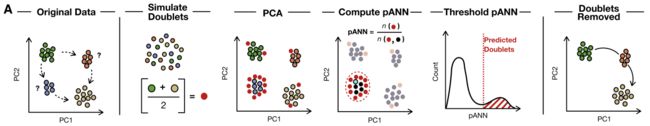
DoubletFinder找doublet的原理也比较简单,看细胞群里面虚拟doublet的占比,超过某个阈值就认定这一群的真实细胞是doublet。在运行DoubletFinder之前,需要对细胞进行初聚类,这和上一种方法是一样的。
library(Seurat)
library(DoubletFinder)
test.seu <- Createtest.seuratObject(counts = test)
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
test.seu <- FindVariableFeatures(test.seu, selection.method = "vst", nfeatures = 2000)
test.seu <- ScaleData(test.seu, features = rownames(test.seu))
test.seu <- RunPCA(test.seu, features = VariableFeatures(test.seu),npcs = 50)
test.seu <- FindNeighbors(test.seu, dims = 1:20)
test.seu <- FindClusters(test.seu, resolution = 0.5)
test.seu <- RunUMAP(test.seu, dims = 1:20)
test.seu <- RunTSNE(test.seu, dims = 1:20)接下来选择一个重要参数pK,这个参数定义了PC的neighborhood size
sweep.res.list <- paramSweep_v3(test.seu, PCs = 1:10, sct = FALSE)
for(i in 1:length(sweep.res.list)){
if(length(sweep.res.list[[i]]$pANN[is.nan(sweep.res.list[[i]]$pANN)]) != 0){
if(i != 1){
sweep.res.list[[i]] <- sweep.res.list[[i - 1]]
}else{
sweep.res.list[[i]] <- sweep.res.list[[i + 1]]
}
}
}
sweep.stats <- summarizeSweep(sweep.res.list, GT = FALSE)
bcmvn <- find.pK(sweep.stats)
pk_v <- as.numeric(as.character(bcmvn$pK))
pk_good <- pk_v[bcmvn$BCmetric==max(bcmvn$BCmetric)]nExp_poi <- round(0.1*length(colnames(test.seu)))指定期望的doublet数
test.seu <- doubletFinder_v3(test.seu, PCs = 1:10, pN = 0.25, pK = pk_good, nExp = nExp_poi, reuse.pANN = FALSE, sct = FALSE)这一行是主要代码,会在[email protected]数据框的基础上加上两列
colnames([email protected])[ncol([email protected])]="DoubletFinder"第二列换一个列名
DF_df <- [email protected][,c("CB","DoubletFinder")]
write.table(DF_df, file = "DoubletFinder_result.txt", quote = FALSE, sep = '\t', row.names = FALSE, col.names = TRUE)
DimPlot(test.seu,reduction = "tsne",pt.size = 2,group.by = "DoubletFinder")最终的效果如下图所示:

3. Scrublet
是一个Python包,安装可以参考:https://github.com/AllonKleinLab/scrublet
>>> import scrublet as scr
>>> import numpy as np
>>> infile = "test.count.txt"
>>> outfile = "Scrublet_result.txt"下面的代码对输入文件做预处理,包括提取出CB,提取count矩阵并转置
>>> finallist = []
>>> with open(infile, 'r') as f:
... header = next(f)
... cell_barcodes = header.rstrip().split('\t')
... for line in f:
... tmpline = line.rstrip().split('\t')[1: ]
... tmplist = [int(s) for s in tmpline]
... finallist.append(tmplist)
>>> finalarray = np.array(finallist)
>>> count_matrix = np.transpose(finalarray)Scrublet检测doublet主要代码如下:
>>> scrub = scr.Scrublet(count_matrix, expected_doublet_rate = 0.1)
>>> doublet_scores, predicted_doublets = scrub.scrub_doublets()
>>> predicted_doublets_final = scrub.call_doublets(threshold = 0.2)第三行换阈值,更新注释结果,接下来保存结果
>>> with open(outfile, 'w') as f:
... f.write('\t'.join(['CB', 'Scrublet', 'Scrublet_Score']) + '\n')
... for i in range(len(doublet_scores)):
... if predicted_doublets_final[i] == 0:
... result = 'Singlet'
... else:
... result = 'Doublet'
... f.write('\t'.join([cell_barcodes[i], result, str(doublet_scores[i])]) + '\n')切换到R环境,在tsne上看看效果
SR_df=read.table("Scrublet_result.txt",header = T,sep = "\t",stringsAsFactors = F)
[email protected]=inner_join([email protected],SR_df,by="CB")
rownames([email protected])[email protected]$CB
DimPlot(test.seu,reduction = "tsne",pt.size = 2,group.by = "Scrublet")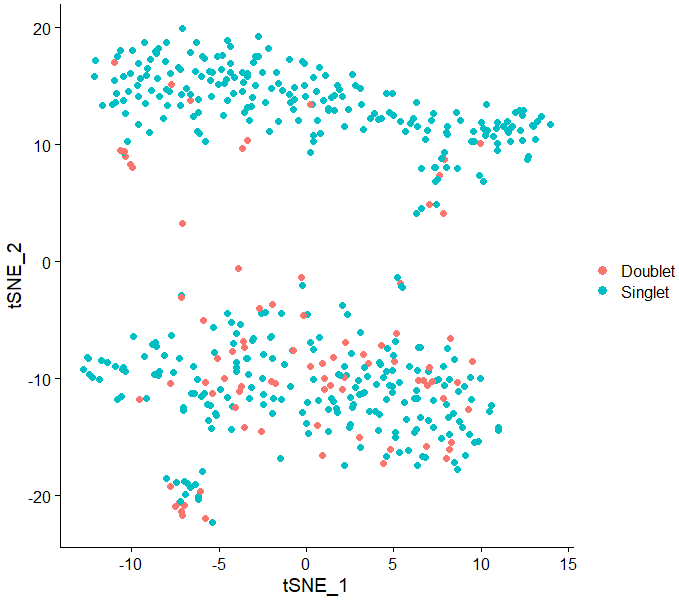
到这里就把三个软件的基本使用讲完了,我只使用了一个实际数据演示,结果并不足以反映这几个软件谁好谁坏,小伙伴们需要结合自己的数据选择合适的软件。开篇提到的文献可信度还是挺高的,大家有需要的话可以认真学一下DoubletFinder这个软件。 另外,可以在github上看到这几个软件是相互推荐的,在生信圈子还挺少见~
因水平有限,有错误的地方,欢迎批评指正!