同源建模MOE:请叫我永远的神!
同源建模
实/操/篇
1.分子力场的设置
MOE中缺省的分子力场MMFF94x是一个用于小分子和复合物模拟的力场,不能很好地适用于大分子模拟,由于这里我们要进行同源模建,模拟对象是蛋白质结构,因此首先要将缺省分子力场MMFF94x更改为适用于蛋白质模拟的相关分子力场,如Amber或者CHARMM等。
具体操作:
执行MOE|WindowIPotential Setup..命令,打开“Potential Setup” 面板;单击“Load.."下拉菜单并选择“Amber99"分子力场,如图1所示。

图1 分子力场设置面板
2.导人目标蛋白序列
具体操作:
执行SE|File|Open...命令,导人目标蛋白序列$MOE/sample/mol/casp_t69.pir;并将“Sequence Editor”窗口与MOE主窗口进行同步,即执行SE|Selection命令,单击选中“Synchronize"复选框,如图2所示。

图2 让“Sequence Editor窗口与MOE主窗口保持同步
3.寻找模板
具体操作:首先执行SE|Homology|PDB Search.命令,如图3所示。

图3 “Sequence Editor”窗口中“PDB Search..."命令
单击“MOE SearchPDB"面板中“Chain右边的数字“1”选项(这里目标蛋白序列只有一条链);再单击“Search"按钮,“MOESearchPDB"面板下面的文本框中将显示出最终序列搜索结果,如图4所示。

图4 导入提问蛋白序列及其搜索结果
从序列搜索结果中挑选一个合适的模板家族,双击该模板家族;在弹出的“MOE-SearchPDB: Load Alignment”面板上单击"Load All"按钮将该模板家族蛋白序列导人到“Sequence Editor”窗口中,如图5所示。

图5 选中的蛋白家族
如图6所示,联配好的模板家族序列都被导人到“SequenceEditor”窗口。由于“Sequence Editor”窗口与MOE主窗口已经同步,并且模板家族中的蛋白不仅有序列信息,还有三维结构信息,因此在MOE主窗口中出现了该模板家族蛋白的三维结构图。

图6 模板家族蛋白序列联配后的MOE主窗口和对应的“SequenceEditor”窗口
注意:尽管模板家族所有蛋白的序列已经联配好,但是此时还没进行相应的三维结构叠合。
具体操作:
选上模板家族蛋白的所有序列(在“SequenceEditor”窗口中先选择目标蛋白序列,然后翻转选择即可,即执行SE|Selection|InvertChains命令);

图7 让“Sequence Editor窗口与MOE主窗口保持同步
接下来将目标蛋白序列对模板家族的所有蛋白序列进行联配,首先打开“MOE-Align”面板,即执行SE|Homology| Align...命令,并单击“freeze""选项(因为模板家族蛋白的序列已经联配好,在接下来目标蛋白序列对模板家族所有序列的联配过程中,模板家族所有序列之间原有的联配保持不变),最后单击“OK"按钮,如图7所示。

图8 模板家族所有蛋白的三维结构叠合
如图8所示,由于在“MOE-Align”面板中的操作包括了对有三维结构信息的蛋白同时进行结构叠合操作,因此MOE主窗口中,所选模板家族蛋白的三维结构也进行了相应的叠合。
打开“SVL Commands"窗口(单击MOE主窗口中的“SVL"按钮) ,观察“pairwise percentage residue identity”,可以发现所选模板家族蛋白中,1PWB与我们的目标蛋白t69的相似分值最高,如图9所示,这些信息将为后面选择单个模板提供参考。

图9 目标序列与模板蛋白所有序列的联配得分
注意:仔细观察上面的数值,A-B(比如t69-1PWB为60.3)与B-A(比如1PWB-t69为58.3)的值并不相同,这是因为尽管计算的时候两者的分子相同,但是由于这两条序列各自的氨基酸数目不同,导致分母不同,最后结果也就不同。
4.根据模板、loop库和边链库构建目标蛋白的三维结构
具体操作:
打开同源模建面板,即执行SE|Homology| Homology Model...命令,对模板、loop库和边链库等进行相关设置(这里仅单击选中“OpenDatabaseViewer"复选框,其他参数保留缺省设置),最后单击“OK”按钮,如图10所示。

图10 “Homology Model"面板参数设置
图11显示的是新生成的目标蛋白质三维结构数据库,其中最后一个模型是对前面三个模型整体平均优化得到的;该结构同时显示在MOE主窗口中,如图12所示。
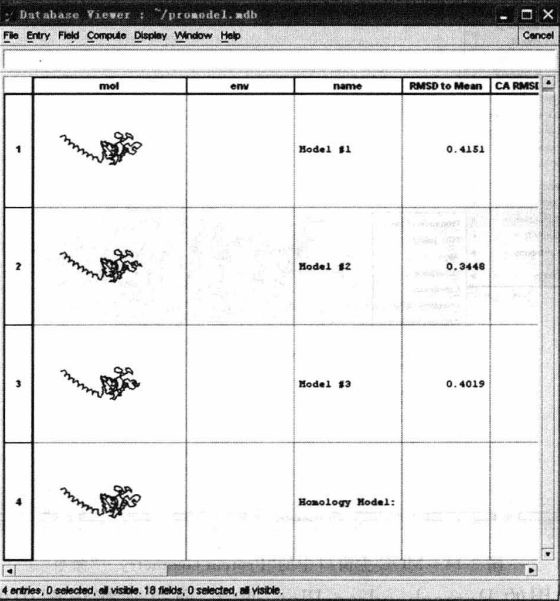
图11 目标蛋白序列对应的三维结构数据库

图12 目标蛋白三维结构
5.目标蛋白结构的评估
具体操作:
执行MOE|Compute|Biopolymer|Protein Geometry...命令,如图13所示。
图13 MOE主窗口中的"Protein Geometry..."命令
图14即是常用的Ramachandran Plot,通常,通过观察该图中的Outlier的个数及所处的位置来判断目前模建得到的蛋白结构是否合理。
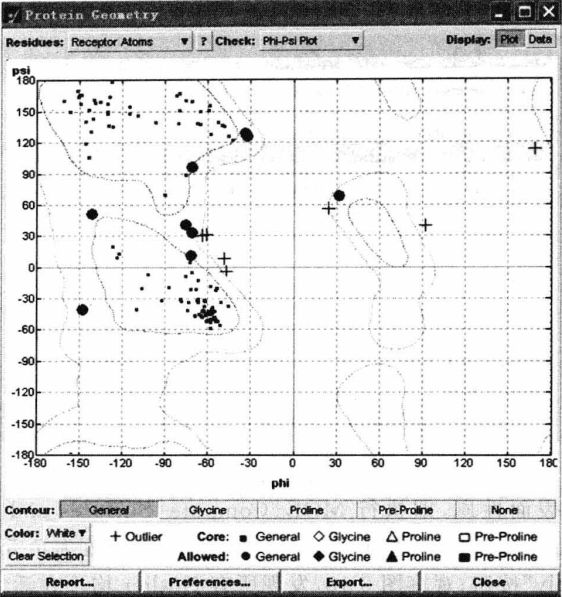
图14 Ramachandran Plot
具体操作:
在“Protein Geometry”面板上选上所有Outlier 按住左键拖动鼠标),则相应在MOE主窗口中这些Outlier也同时被选上;用Space Filling显示这些Outlier, 即在MOE主窗口中执行RHS|Mode| Space Filling命令,如图15所示。

图15 突出显示Outlier位置
判定这些Outlier是否处于活性位点中。首先应获得目标蛋白的活性位点,即执行MOE|Compute|SiteFinder..命令,单击“Apply"按钮;然后选择第一个位点作为活性位点,并通过单击“Site Finder”面板中“Dummies...”按钮获得相关假原子,如图16所示。

图16 获取蛋白活性位点
计算活性位点表面性质,即执行MOE|Compute| Surfaces and Maps...命令,并在“Surfaces and Maps”面板中进行相关设置(这里仅将距离更改为7.5埃,保留其他缺省设置),然后单击“Apply”按钮;观察图17,发现所有Outlier均不在活性位点内,因此这个模型可以保存下来用于后续的研究。
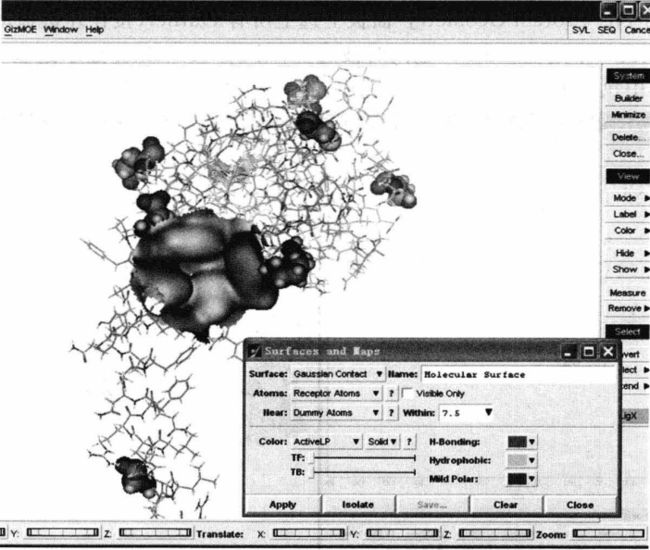
图17 观察Outlier与活性位点的位置关系
6.目标蛋白结构的再优化或重新模建
可以按照如下顺序对评估不合格的目标蛋白结构进行再优化或者重新模建:
①如图18所示对整个蛋白或者对所选残基(比如Outlier或者包含Outlier周边残基一起)进行特定或进一步的能量优化。
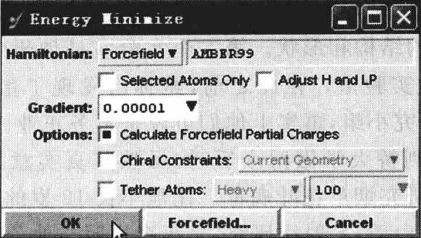
图18 “Energy Minimize"面板设置
具体操作:
执行MOE| Compute | Energy Minimize..命令,根据具体需要在“EnergyMinimize"面板中进行相应的参数调整。

图19 将分子力场更换为CHARMM后模建得到的结果
②如图19所示,选择另外一种分子力场重复步骤(1)的操作(这里将Amber更换为CHARMM)。
③按照前面两个步骤还是得不到更合理的模型,则可以考虑从模板家族中选择另外的蛋白作为模板;如果还不行,则进一步重新选择模板家族,直到能够得到好的模型为止。
近期课程(惊喜福利):
CADD-Rosetta(线下)-NAMD-AMBER药物及分子动力学专题
专题一:Rosetta从头蛋白抗体设计应用(线下)
专题二:NAMD分子动力学模拟在生物及材料计算中的应用——
赠送Gromacs专题(三天)往期直播课录播视频回放。
专题三:AMBER分子动力学能量优化与分析、结合自由能计算——
赠送Gromacs专题(三天)、薛定谔专题(两天)任选其一的往期直播课程录播视频回放。
专题四:CADD蛋白结构分析、虚拟筛选、分子对接——
报名后赠送AIDD专题(五天)、Gromacs专题(三天)、薛定谔专题(两天)任选其一的往期直播课程录播视频回放
大纲,报名联系人等详细内容请查阅原文,原文链接:
计算机辅助药物设计及分子动力学技术与应用2023.7腾讯文档-在线PDF https://docs.qq.com/pdf/DTXplYkZVWGtMUndh
https://docs.qq.com/pdf/DTXplYkZVWGtMUndh
专题五:代谢组学及网络药理学研究技术与实践(录播)
大纲,报名联系人等详细内容请查阅原文,原文链接:代谢组学数据分析及网络药理学研究技术与实践录播腾讯文档-在线PDFhttps://docs.qq.com/pdf/DTUhxelp0U2NvT1ho
征稿专栏
征稿啦!!!“计算机辅助药物设计CADD”微信公众号自创办以来得到了广大科研工作者和研究生的广泛关注和支持。为更好地服务计算机辅助药物设计研究和应用,本公众号现因业务需要长期招聘供稿作者。
投稿到[email protected],邮件主题请注明“姓名+供稿作者”。
欢迎药物设计类相关专业的科研爱好者加入“计算机辅助药物设计CADD”团队。