复现Nature图表:GSEA分析及可视化包装函数
这篇帖子主要的目的是写一个转录组GSEA分析和可视化通用的函数。起因是我们想要复现一篇文章的GSEA可视化图片,这个Nature文章GSEA可视化挺好的:
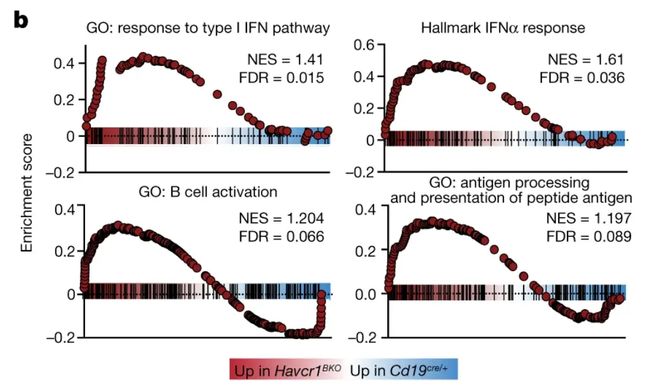
image.png
(reference:B-cell-specific checkpoint molecules that regulate anti-tumour immunity)后来干脆一不做二不休,写一个函数吧,有差异分析结果即可,可视化也是一键出结果。算是懒人福音吧。我们此部分一共有两个函数,一个是KS_GSEA,作用是进行转录组数据GSEA分析,提供clusterProfiler和fgsea两种R包分析方式,KS_GSEA适用于human和mouse两个物种,支持KEGG、GO(BP)的GSEA分析。KS_GSEA_plot作用是进行GSEA结果的可视化,可选择需要的通路进行展示。
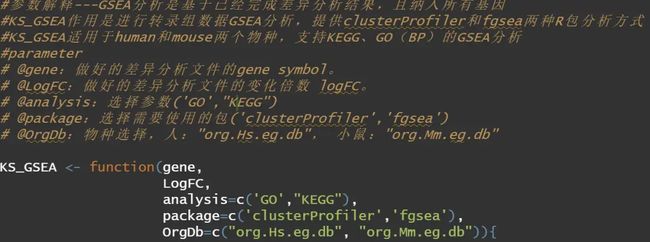
image.png
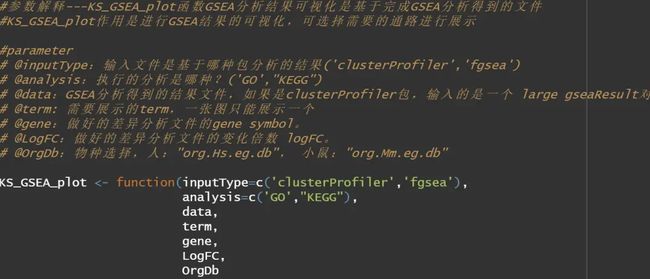
image.png
这里我们以一个单细胞的数据为例,首先做一下差异分析。Bulk RNA的差异分析不再多说。GSEA分析是基于已经完成差异分析结果,且纳入所有基因。目前R做GSEA用的比较多的是clusterProfiler和fgsea包,所以这两种包的分析方式我们都包含进去了。使用的时候选择自己需要的即可。
library(Seurat)
library(msigdbr)
library(fgsea)
library(clusterProfiler)
library(ggplot2)
#加载函数
source('./KS_GSEA.R')
source('./KS_GSEA_plot.R')
#差异基因-----------------------------------------------------------------------
load("D:/KS项目/公众号文章/GSEA分析及可视化函数/sce_mar.RData")
df <- FindMarkers(Macrophage, min.pct = 0, logfc.threshold = 0,
group.by = "group",ident.1 ="BM",ident.2="GM")
df$gene = rownames(df)
得到的差异分析结果是一个数据框,包含gene symbol这一列,还有logFC这一列。我们知道GSEA分析需要对基因按照FC进行排序,我们这里的分析不需要事先进行排序,只要提供gene和logFC即可。两种R包的结果我们都做一下。
#GSEA分析-----------------------------------------------------------------------
GSEA_CP <- KS_GSEA(gene = df$gene,
LogFC = df$avg_log2FC,
analysis = "KEGG",
package = 'clusterProfiler',
OrgDb = 'org.Hs.eg.db')
class(GSEA_CP)
# [1] "gseaResult"
# attr(,"package")
# [1] "DOSE"
GSEA_F <- KS_GSEA(gene = df$gene,
LogFC = df$avg_log2FC,
analysis = "KEGG",
package = 'fgsea',
OrgDb = 'org.Hs.eg.db')
class(GSEA_F)
# [1] "data.table" "data.frame"
可以看到,clusterProfiler包返回的是一个gseaResult,fgsea包返回的是一个"data.table","data.frame"。这些结果就是我们下一步可视化的输入文件。运行结束后,分析结果已txt或者csv的形式直接保存到当前环境路径下!
image.png
我们对可视化函数进行了设置,NES>0的结果点用红色显示。NES<0的结果用蓝色点显示。这里我们挑选自己感兴趣的通路进行可视化。
#NES>0
p1=KS_GSEA_plot(inputType = "clusterProfiler",
analysis = "KEGG",
data = GSEA_CP,
term = 'Oxidative phosphorylation',
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
p2=KS_GSEA_plot(inputType = "fgsea",
analysis = "KEGG",
data = GSEA_F,
term = 'Huntingtons disease',
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
p1+p2
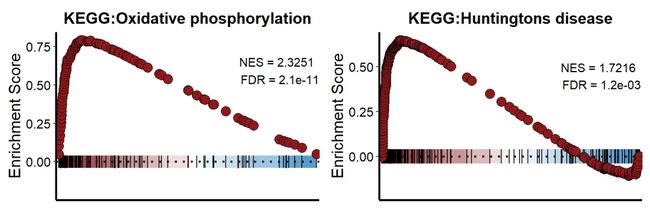
image.png
NES<0,这里需要注意一个问题,clusterProfiler和fgsea虽然都是GSEA分析,但是两者得到的结果并不是一摸一样完全相同的,总是有一些出入,这是因为数据库,分析方式不一样。选择需要的包使用一个即可。
#NES<0
p3=KS_GSEA_plot(inputType = "clusterProfiler",
analysis = "KEGG",
data = GSEA_CP,
term = 'JAK-STAT signaling pathway',
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
p4=KS_GSEA_plot(inputType = "fgsea",
analysis = "KEGG",
data = GSEA_F,
term = 'Jak stat signaling pathway',
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
p3+p4
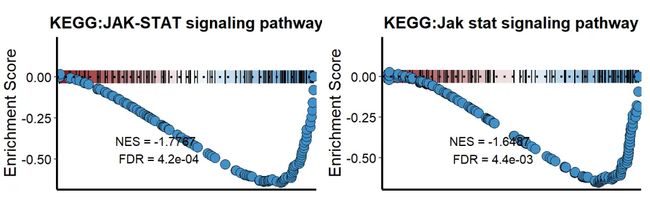
image.png
当然了,我们可以利用循环一次性可视化通路。
#批量循环
#NES>0
pathway <- c('Oxidative phosphorylation',
"Parkinson disease",
"Biosynthesis of amino acids",
"Cardiac muscle contraction")
pathway_list <- list()
for (i in 1:length(pathway)) {
p = KS_GSEA_plot(inputType = "clusterProfiler",
analysis = "KEGG",
data = GSEA_CP,
term = pathway[i],
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
pathway_list[[i]] <- p
}
#组合图
CombinePlots(pathway_list, ncol = 2)
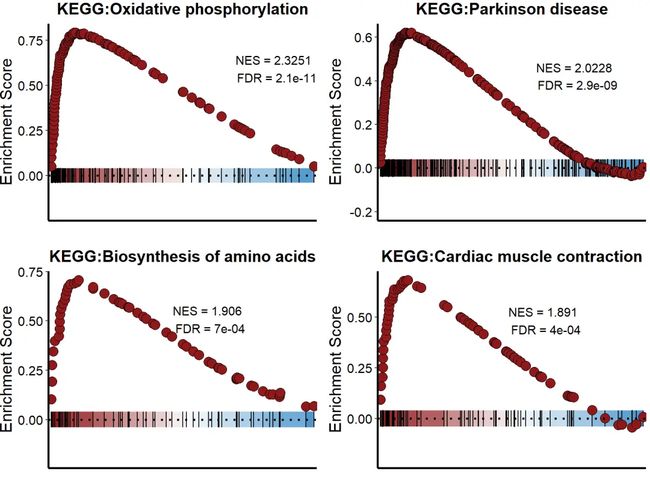
image.png
用另一个数据批量做一下下调的。
#NES<0
pathway1 <- c('Melanoma',
'Prostate cancer',
'Ecm receptor interaction',
'Tgf beta signaling pathway')
pathway_list1 <- list()
for (i in 1:length(pathway)) {
p = KS_GSEA_plot(inputType = "fgsea",
analysis = "KEGG",
data = GSEA_F,
term = pathway1[i],
gene = df$gene,
LogFC = df$avg_log2FC,
OrgDb = 'org.Hs.eg.db')
pathway_list1[[i]] <- p
}
#组合图
CombinePlots(pathway_list1, ncol = 2)
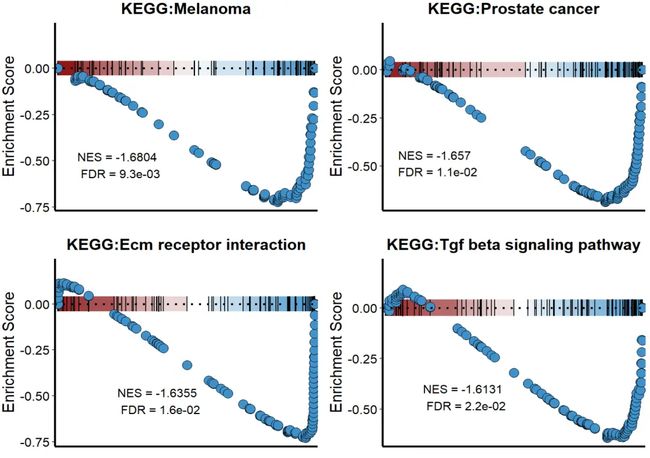
image.png
这就是所有内容了,希望对你有所启发。这个函数其实并不是很完美,首先是物种只支持小鼠和人,其次是分析参数我没有再设置,用的是我常用的。当然了,这个函数框架我提供了,需要更加个性化的可以自行修改。更多精彩请至我们的公众号---KS科研分享与服务