孟德尔随机化,其实MR自带循环
关注微信公众号CodeMed
在进行孟德尔随机化研究中,尤其是目前组学当道,大家都纷纷加入,看已发表的文章,想把其他人的研究结局,替换成自己的的研究方向,进行探索(发SCI)...
但是,不可避免的出现一个问题:我R基础不行,不会写循环啊......
---- 其实TwoSampleMR包自带循环
01
入门级做法
常规认知是如同TwoSampleMR Git Page官方文档提供的代码进行操作
例如我们要研究 HDL, LDL and Total cholesterol与coronary heart disease (CHD),首先肯定会做单因素。常规会像下面代码,进行三次重复操作
1. 先做HDL的
# 提取研究暴露的工具变量bmi_exp_dat <- extract_instruments(outcomes = 'ieu-a-299')# 根据暴露的SNP, 提取研究结局的chd_out_dat <- extract_outcome_data(snps = bmi_exp_dat$SNP, outcomes = 'ieu-a-7')#dat <- harmonise_data(bmi_exp_dat, chd_out_dat)# 后续一系列MR和敏感性分析mr(dat)
2. 在做LDL的
# 提取研究暴露的工具变量bmi_exp_dat <- extract_instruments(outcomes = 'ieu-a-300')# 根据暴露的SNP, 提取研究结局的chd_out_dat <- extract_outcome_data(snps = bmi_exp_dat$SNP, outcomes = 'ieu-a-7')#dat <- harmonise_data(bmi_exp_dat, chd_out_dat)# 后续一系列MR和敏感性分析mr(dat)
3. 在做Total cholesterol
# 提取研究暴露的工具变量bmi_exp_dat <- extract_instruments(outcomes = 'ieu-a-302')# 根据暴露的SNP, 提取研究结局的chd_out_dat <- extract_outcome_data(snps = bmi_exp_dat$SNP, outcomes = 'ieu-a-7')#dat <- harmonise_data(bmi_exp_dat, chd_out_dat)# 后续一系列MR和敏感性分析mr(dat)
...
如果还有其他变量,例如微生物组,蛋白组,代谢组...这些变量可不止3个,单独拿大家用的最多的菌群就有211个分类群,岂不是...

我又不会写什么for循环,封装什么Function
02
TSMR自带循环
接下来看下TwoSampleMR自带循环
大家可以复制下面代码直接放到R执行
library(TwoSampleMR)# 自动一次提取三个变量的工具变量,只需要把id填入Multiple_exp_dat <- extract_instruments(outcomes = c("ieu-a-299", "ieu-a-300", "ieu-a-302"))# 提取三个变量合并SNP的结局# 当然这边也可以填入多个研究结局 outcomes=c("ieu-a-2","ieu-a-7")outcome <- extract_outcome_data(snps = Multiple_exp_dat$SNP,outcomes = "ieu-a-2")# harmonise # 【以这个函数,查看原理】Multiple_har <- harmonise_data(Multiple_exp_dat,outcome)# 进行mr分析Multiple_res <- mr(Multiple_har)# 进行过滤Multiple_har=steiger_filtering(Multiple_har)# 进行MRPRESSOres_MRPRESSO <- run_mr_presso(dat = Multiple_har)# 进行异质性heterogeneity <- mr_heterogeneity(Multiple_har)# 进行水平多效性pleiotropy <- mr_pleiotropy_test(Multiple_har)
上面结果大家可以看到,R的Console输出的信息,自动进行了一个个单因素分析。
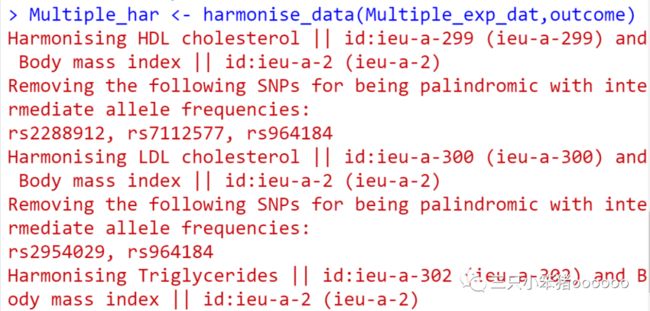

查看Multiple_res,可以看到三份结果自动合并在一起了
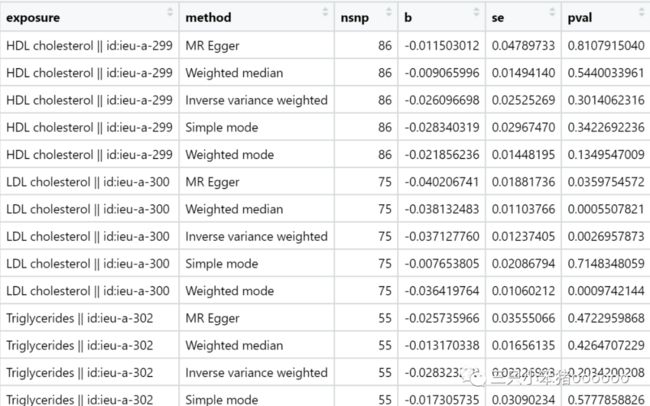
TwoSampleMR自带的函数会自动根据id.exposure和id.outcome进行配套分析,就像你一个个分析HDL, LDL and Total cholesterol然后在合并起来的结果是一样的。
本地Summary GWAS数据导入,也会自动生成唯一的id.exposure与id.outcome
最后我们再来看下代码的原理,来验证下为什么说TwoSampleMR自带循环
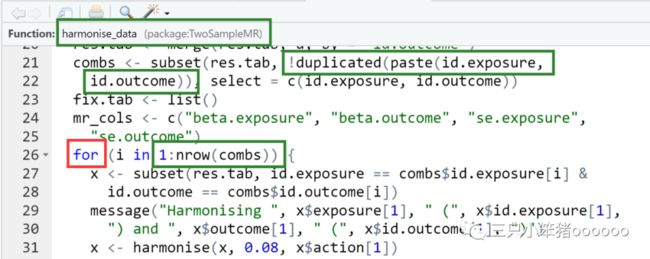
以harmonise_data函数为例(选中函数,按F2)可以查看源码
subset(res.tab, !duplicated(paste(id.exposure, id.outcome)), select = c(id.exposure, id.outcome)) 提取配对的单因素
combs为有多少对id.exposure和id.exposure进行for循环分析
-
# combs的数据提取出来例如为下面数据框# 3个暴露,2个结局> combs <- data.frame(expID=c("A1","A2","A3","A1","A2","A3"),> outID=c("B1","B1","B1","B2","B2","B2"))> 1:nrow(combs)[1] 1 2 3 4 5 6