Modeller多模板同源建模教程
Modeller多模板同源建模教程
1. Modeller简介
Modeller用于蛋白质三维结构的同源或比对建模。用户可根据氨基酸序列自动计算出一个包含所有非氢原子排列的三维结构模型。Modeller通过满足空间约束实现比较蛋白质结构建模,并可以执行许多附加任务:包括蛋白质结构环的从头建模、根据灵活定义的目标函数优化各种蛋白质结构模型、蛋白质序列的多重排列和结构聚类、序列数据库搜索、蛋白质结构比较等。Modeller可适用于大多数Unix/Linux系统、Windows和Mac。

2. Modeller下载安装
| Anaconda Python (“conda”) | |
|---|---|
| Windows (32-bit) | Installation guide |
| Windows (64-bit) | Installation guide |
| Mac (32-bit or 64-bit Intel) | Installation guide |
| Linux (32-bit RPM) | Installation guide |
| Linux (64-bit x86_64 RPM) | Installation guide |
| Linux (32-bit Debian/Ubuntu package) | Installation guide |
| Linux (64-bit x86_64) | Installation guide |
| Generic Unix tarball | Installation guide |
| 推荐使用Conda安装,安装命令如下: |
conda config --add channels salilab
conda install modeller
安装后,系统将提示添加Modeller许可证密钥。密钥可通过教育邮箱.edu注册申请获得。编辑/Library/modeller-9.24/modlib/modeller/config.py 文件,修改密钥。
3. Modeller多模板建模流程
参考自官方教程Advanced example
3.1 下载fasta文件
- Uniprot下载(浏览器下载)
例: https://www.uniprot.org/uniprot/P29590.fasta
- wget下载(shell下载)
wget https://www.uniprot.org/uniprot/P29590.fasta
3.2 序列比对查找模板
-
使用SWISS-MODEL进行模板比对

填写序列并搜索模板 -
使用BlastP进行模板比对

根据
同源性和cover位置选择多个模板

3.3 多模板align
由Modeller中的salign.py脚本生成多模板的聚类对齐。
mod9.24 salign.py
salign.py脚本如下:
# Illustrates the SALIGN multiple structure/sequence alignment
from modeller import *
log.verbose()
env = environ()
env.io.atom_files_directory = './'
aln = alignment(env)
for (code, chain) in (('2mdh', 'A'), ('1bdm', 'A'), ('1b8p', 'A')):
mdl = model(env, file=code, model_segment=('FIRST:'+chain, 'LAST:'+chain))
aln.append_model(mdl, atom_files=code, align_codes=code+chain)
for (weights, write_fit, whole) in (((1., 0., 0., 0., 1., 0.), False, True),
((1., 0.5, 1., 1., 1., 0.), False, True),
((1., 1., 1., 1., 1., 0.), True, False)):
aln.salign(rms_cutoff=3.5, normalize_pp_scores=False,
rr_file='$(LIB)/as1.sim.mat', overhang=30,
gap_penalties_1d=(-450, -50),
gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0,
dendrogram_file='fm00495.tree',
alignment_type='tree', # If 'progresive', the tree is not
# computed and all structues will be
# aligned sequentially to the first
feature_weights=weights, # For a multiple sequence alignment only
# the first feature needs to be non-zero
improve_alignment=True, fit=True, write_fit=write_fit,
write_whole_pdb=whole, output='ALIGNMENT QUALITY')
aln.write(file='fm00495.pap', alignment_format='PAP')
aln.write(file='fm00495.ali', alignment_format='PIR')
aln.salign(rms_cutoff=1.0, normalize_pp_scores=False,
rr_file='$(LIB)/as1.sim.mat', overhang=30,
gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3),
gap_gap_score=0, gap_residue_score=0, dendrogram_file='1is3A.tree',
alignment_type='progressive', feature_weights=[0]*6,
improve_alignment=False, fit=False, write_fit=True,
write_whole_pdb=False, output='QUALITY')
只需修改第8行中的PDB ID和Chain
for (code, chain) in ((‘2mdh’, ‘A’), (‘1bdm’, ‘A’), (‘1b8p’, ‘A’)):
3.4 修改查询序列文件
将下载的P29590.fasta文件修改为P29590.ali文件如下:
>P1;P29590
sequence:P29590:::::::0.00: 0.00
MEPAPARSPRPQQDPARPQEPTMPPPETPSEGRQPSPSPSPTERAPASEEEFQFLRCQQC
QAEAKCPKLLPCLHTLCSGCLEASGMQCPICQAPWPLGADTPALDNVFFESLQRRLSVYR
DAMTQALQEQDSAFGAVHAQMHAAVGQLGRARAETEELIRERVRQVVAHVRAQERELLEA
VDARYQRDYEEMASRLGRLDAVLQRIRTGSALVQRMKCYASDQEVLDMHGFLRQALCRLR
QEEPQSLQAAVRTDGFDEFKVRLQDLSSCITQGKDAAVSKKASPEAASTPRDPIDVDLPE
FNLQALGTYFEGLLEGPALARAEGVSTPLAGRGLAERASQQS*
第一行包含序列码,格式为>P1;code。
第二行有10个用冒号分隔的字段,如果适用,通常包含有关结构文件的信息。其中只有两个字段用于序列,sequence表示文件包含没有已知结构的序列,P29590表示序列的文件名。
文件的其余部分包含P29590的序列,并用*标记其结尾。
使用标准的单字母氨基酸代码。注意,它们必须是大写字母;一些小写字母用于非标准残基,请参阅modlib/restyp.lib文件中的有关详细信息。
3.5 查询序列与模板align
由Modeller中的align2d_mult.py脚本生成查询序列与模板序列的对齐。
mod9.24 align2d_mult.py
align2d_mult.py脚本如下:
from modeller import *
log.verbose()
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib')
# Read aligned structure(s):
aln = alignment(env)
aln.append(file='fm00495.ali', align_codes='all')
aln_block = len(aln)
# Read aligned sequence(s):
aln.append(file='TvLDH.ali', align_codes='TvLDH')
# Structure sensitive variable gap penalty sequence-sequence alignment:
aln.salign(output='', max_gap_length=20,
gap_function=True, # to use structure-dependent gap penalty
alignment_type='PAIRWISE', align_block=aln_block,
feature_weights=(1., 0., 0., 0., 0., 0.), overhang=0,
gap_penalties_1d=(-450, 0),
gap_penalties_2d=(0.35, 1.2, 0.9, 1.2, 0.6, 8.6, 1.2, 0., 0.),
similarity_flag=True)
aln.write(file='TvLDH-mult.ali', alignment_format='PIR')
aln.write(file='TvLDH-mult.pap', alignment_format='PAP')
修改替换TvLDH为自己的P29590.ali中的名字P29590
sed -i 's\TvLDH\P29590\g' align2d_mult.py
3.6 多模板建模
由Modeller中的model_mult.py脚本生成建模结构。(生成100个结构)
mod9.24 model_mult.py
model_mult.py脚本如下:
from modeller import *
from modeller.automodel import *
env = environ()
a = automodel(env, alnfile='TvLDH-mult.ali',
knowns=('1bdmA','2mdhA','1b8pA'),
sequence='TvLDH',
assess_methods=(assess.DOPE,assess.GA341))
a.starting_model = 1
a.ending_model = 100
a.make()
修改TvLDH为自己的P29590.ali中的名字P29590
sed -i 's\TvLDH\P29590\g' model_mult.py
修改第六行中的PDB ID和Chain
最后根据model_mult.log文件中的DOPE(越小越好)、molpdf(越小越好)、GA341(越小越好)分值确定最终结构。
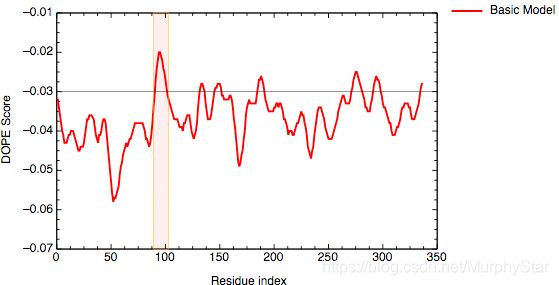
3.7 DOPE评价
由Modeller中的evaluate_model.py脚本评价建模结构。
mod9.24 evaluate_model.py
evaluate_model.py脚本如下:
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file
mdl = complete_pdb(env, 'TvLDH.B99990001.pdb')
# Assess all atoms with DOPE:
s = selection(mdl)
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='TvLDH.profile',
normalize_profile=True, smoothing_window=15)
修改TvLDH为序列名字P29590
sed -i 's\TvLDH\P29590\g' evaluate_model.py
可根据可以使用shell自带的gnuplot作图,代码如下:
gnuplot <<EOF
set term pngcairo
set output "TvLDH.png"
set xlabel 'Residue index'
set ylabel 'DOPE Score'
plot "TvLDH.profile" using 1:42 with lines
set output
EOF