5+单细胞+脂质代谢+预后模型+实验

肝癌的死亡率在所有癌症中排名第三高,并且是最常见的第六种癌症(1)。肝细胞癌(HCC)是一种起源于肝细胞的肿瘤。HCC是原发性肝癌(PLC)的主要病理类型,占所有PLC实例的75-85%(2)。绝大多数HCC都是由慢性疾病引起的,其中大部分病例据报道是由慢性肝病演变而来。这主要是因为病毒感染,包括乙型肝炎病毒(HBV)和丙型肝炎病毒(HCV),以及酗酒(3)。根据巴塞罗那肝癌诊所(BCLC)分期系统,建议早期诊断的HCC患者接受手术切除、肝移植和局部切除(射频消融)治疗。中期患者通常接受经动脉化疗栓塞(TACE)治疗,而系统性治疗主要用于晚期患者。晚期患者通常有症状,尽管他们的肝功能有一定程度的受损(4, 5)。值得注意的是,对于处于晚期的患者来说,很少或没有可用的治疗方法可以提高生存率。
1.单细胞基因表达谱揭示了原发性肝细胞癌肿瘤TME中的六种主要细胞类型
作者根据细胞的表达谱进行了降序和无监督的细胞聚类,以识别细胞类型。原始数据集使用Seurat软件包进行读取。然后,使用以下标准对基因和细胞进行了初步筛选:一个基因必须在至少3个细胞中表达,并且在该细胞中至少测量到200个基因。接下来进行了进一步的质量控制,提取具有>200个和<8000个表达基因以及<30%线粒体基因的细胞。然后,对数据进行了归一化处理,得到了2000个高变异基因用于后续的降维处理。去除细胞周期效应后,得到了一个包含58,475个细胞和24,746个基因的表达矩阵。接下来,作者使用已知的标记基因来定义广泛的细胞类别,并获得了以下六个主要的细胞亚群:T/NK细胞、肝细胞、巨噬细胞、内皮细胞、成纤维细胞和B细胞(图1A、B)。来自不同患者的肿瘤和正常癌旁组织的细胞被分类为六个类别(图1C)。由于增殖是肿瘤细胞的一个特征,作者采用了细胞周期评分方法来分析细胞周期。这张图片显示的结果表明,大部分细胞处于G1期,只有少数细胞处于G2/M期和S期(图1D)。

图1 从8个原发性肝癌病灶及其相邻样本中分离的单个细胞的特征
2.与葡萄糖和脂质代谢途径相关的基因在肝细胞中上调表达
为了探索不同细胞亚群之间的通路异质性存在,作者使用特征基因进行了通路活性和GSEA分析。在肝细胞亚群中,与癌症相关的许多通路被上调,包括氧化磷酸化、糖酵解以及脂肪酸、胆汁酸和外源物代谢(图2A)。接下来,作者使用scMetabolism软件包计算了每个细胞中每个代谢通路的得分。作者发现上皮细胞亚群富集了大多数代谢通路,主要是调节丙酮酸代谢、柠檬酸三羧酸循环以及三酰甘油、丙酮酸、脂质、碳水化合物、氨基酸及其衍生物、酮体、葡萄糖和脂肪酸以及FoxO介导的氧化应激通路(图2B)。葡萄糖代谢和脂质代谢通路的基因在上皮细胞中也被上调(图2C、D)。为了确定肝上皮细胞在肿瘤和癌旁组织之间代谢通路的差异,作者提取了一个单独的肝细胞亚群并分析了代谢通路的富集情况。引人注目的是,作者发现肿瘤细胞和癌旁细胞的葡萄糖代谢和脂质代谢途径呈相反的趋势(图2E)。因此,作者对这个细胞亚群进行了更深入的分析。
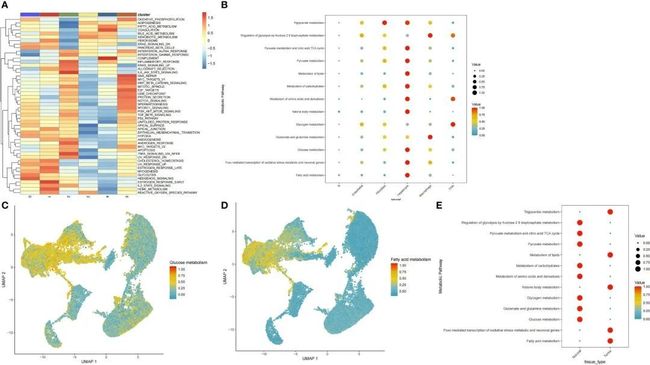
图2 细胞亚群中葡萄糖和脂质代谢途径的分布
3.肿瘤与正常癌旁组织之间的细胞间通讯模式
作者构建了一个肿瘤样本和正常癌旁样本之间的通信网络,以表征信号通路的改变(图3A)。在肿瘤和正常癌旁组织中,分别鉴定出了642个和499个显著的配体-受体(LR)相互作用。图3B展示了肿瘤和正常样本之间所有细胞群体之间通信数量的差异。总的来说,肿瘤样本展示了比其正常对照更多的细胞相互作用,这种现象在整体信号模式中更加明显。接下来,作者研究了这六种细胞类型之间以及特定分子对之间的可能的输出和输入信号。作者发现,无论是输出信号还是输入信号,肿瘤样本始终比正常样本具有更多的信号对。肿瘤样本特有的潜在信号通路包括SPP1、VTN、OCLN、CD46、GDF、EPHA、AGRN、PERIOSTIN和HSPG。在正常样本中,内皮细胞和T/NK细胞是主要的信号提供者和受体,而在肿瘤样本中,成纤维细胞和巨噬细胞分别代表主要的信号提供者和受体(图3C,D)。肿瘤样本和正常样本来源的细胞之间的整体通信概率显著不同。在正常样本来源的细胞间通信的配体受体中,多个通路参与炎症和免疫反应,包括涉及MHC-I、MHC-II、CXCL、补体、CCL和TNF的通路。在肿瘤样本中,细胞间相互作用主要活跃在涉及SPP1、VTN、NOTCH、THY1和CD46的信号通路中(图3E)。为了进一步阐明肝细胞与其他细胞亚群之间的关系,作者生成了一个差异数量和强度的网络图。作者发现肝细胞与内皮细胞和成纤维细胞之间的相互作用显著增强,但与免疫细胞的关联较弱(图3F-G)。
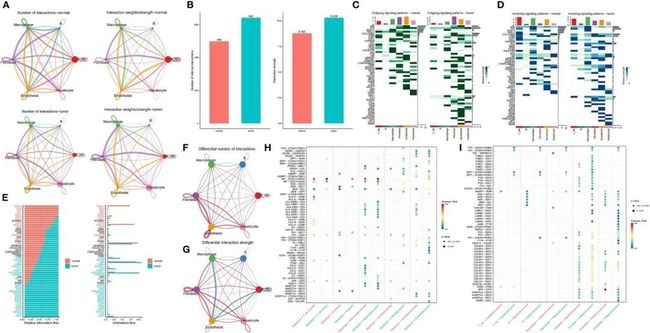
图3 肿瘤和相邻正常组织样本之间细胞相互作用的比较
肝细胞和其他细胞类型中所有配体-受体对的差异分析显示,肿瘤和相邻正常组织之间存在显著不同的模式(图3H-I)。研究表明,CD74通过与MIF相互作用促进肿瘤细胞生长(26)。值得注意的是,肝细胞与T/NK细胞和巨噬细胞之间的MIF-(CD74 + CD44)信号传导介导了免疫抑制效应,这已经被证明促进了癌症进展(27)。阻断MIF-CD74信号不仅抑制了HCC细胞的增殖,还具有抗肿瘤效应。因此,MIF/CD74轴的抑制可能是HCC的有效治疗方法(28)。SPP1编码骨桥蛋白(OPN),一种在与人类疾病相关的各种组织和细胞中表达的磷酸化糖蛋白(29, 30)。值得注意的是,OPN在肿瘤进展中起着关键作用,包括HCC转移和预后,因为它推动了肿瘤细胞在肿瘤微环境中的进化适应。引人注目的是,在肿瘤样本中,SPP1-CD44信号在肝细胞与T/NK细胞、巨噬细胞和成纤维细胞之间存在,但在癌症附近的正常样本中却不存在,进一步支持了SPP1在肿瘤生态系统中的关键作用。
4.肝细胞在肝癌中的转录组异质性
尽管之前存在批次效应,肿瘤细胞仍然显示出特定于患者的表达模式。这表明存在高度的异质性,可能是由于拷贝数变异引起的。在整个恶性和正常肝细胞重新聚类后,确定了六个主要的细胞亚群(图4A)。此外,UMAP图显示出与样本来源相对应的恶性细胞的明显聚类(图4B)。图4C展示了每个细胞亚群的标记基因。接下来,使用irGSEA软件包进行单细胞RNA测序基因集富集分析,发现这些亚群具有独特的激活信号。这些信号包括Hedgehog信号通路(亚群0)、早期雌激素反应(亚群1)、IL6/STAT3和TNF信号通路(亚群2)、外源物代谢和活性氧信号通路(亚群3)以及KRAS信号通路(亚群4)。此外,亚群体5中多个与细胞增殖相关的途径被上调;这些途径包括涉及MYC靶点V1和V2、G2M检查点、E2F靶点、WNT信号和P53靶点的途径(图4D)。活化的KRAS是癌症干细胞(CSC)增殖和肿瘤转移的主要驱动因素(31)。本研究的结果显示,KRAS信号通路在亚群体4中显著上调,并且CSC的标记基因也分布在这个亚群体中(图4E)。
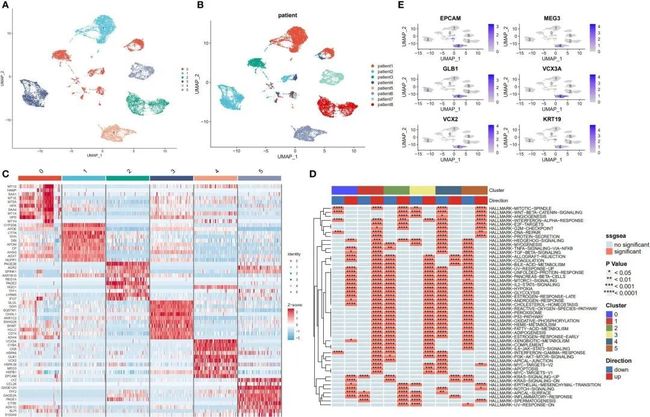
图4 肝细胞癌组织中恶性细胞的转录组异质性
5.肝细胞亚群染色体拷贝数变异的特征
接下来,作者根据转录组数据确定了每个样本中染色体的拷贝数变异,以了解上皮细胞亚群的恶性程度。这张图片显示了相邻正常上皮细胞亚群(对照样本)和肿瘤上皮细胞中低和高的拷贝数变异结果。染色体扩增主要发生在染色体1、3、5、6、7、8、12、15、17、20、21和22中,而缺失最常见于染色体4、9、10、11、13、14、16和18(图5A)。首先,根据每个样本中所有基因的平方和计算拷贝值(拷贝数变异值)。接下来,作者对肿瘤细胞的拷贝数变异值进行排名,以前5%作为参考,并计算其他上皮细胞与参考细胞之间的相关系数。通过设置拷贝数变异值大于0.2和相关系数大于0.2的阈值,确定了肿瘤细胞。以拷贝数变异值为横坐标,相关系数为纵坐标,黑色点代表肿瘤细胞,蓝色点代表正常细胞(图5B)。最后,在UMAP图上确定了13,502个肿瘤细胞和1,718个正常细胞(图5C)。
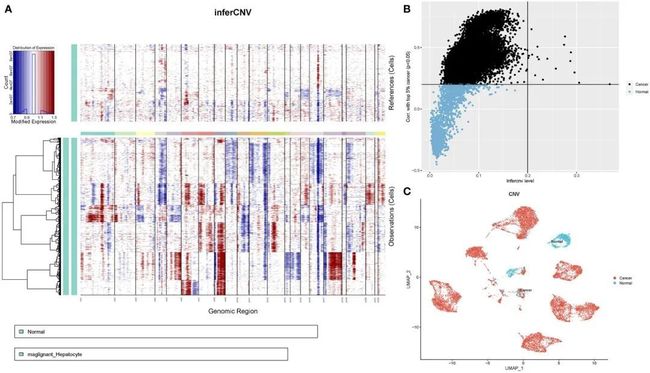
图5 HCC患者上皮细胞的CNV分析
此后,作者使用了FindAllMarkers函数,并设置了筛选条件logfc = 0.25(差异倍数),min. pct = 0.25(最小差异基因表达比率)和pct. diff >0.1(pct.1-pct.2),以识别肝恶性和正常上皮细胞亚群中的标记基因。结果显示共有564个标记基因。作者假设它们在肝细胞癌中的功能与正常上皮细胞有所不同,尽管还需要进一步的研究探索。
6.恶性肝细胞亚群与肝癌预后相关
接下来,作者探讨了肝细胞亚群在肝细胞癌(HCC)患者中的预后作用。通过对TCGA数据库中HCC样本的mRNA表达数据进行分析,作者发现了2900个差异表达基因(图6A)。肝细胞亚群的恶性和非恶性细胞的标记基因与TCGA数据库中与HCC发展相关的差异表达基因相交。值得注意的是,2900个差异表达基因与564个标记基因重叠,从而在HCC中得到了203个差异表达标记基因。随后,这些基因被命名为肝细胞差异基因(HDGs)(图6B)。单变量Cox回归分析显示,101个差异表达标记基因与HCC患者的预后显著相关。为了获得更可靠的预后概况,作者采用了LASSO回归算法进行10倍交叉验证,最小lambda值为0。06321515用于指定一个由11个基因组成的预测模型,即MARCKSL1(类似MARCKS的蛋白1)、SPP1(分泌性磷酸蛋白1)、BSG(Basigin,也称为CD147或EMMPRIN)、CCT3(含TCP1亚单位的分子伴侣蛋白3)、LAGE3(L抗原家族成员3)、KPNA2(核转运蛋白亚单位α2)、SF3B4(剪接因子3b亚单位4)、GTPBP4(GTP结合蛋白4)、PON1(对硫磷酸酯酶1)、CFHR3(补体因子H相关蛋白3)和CYP2C9(细胞色素P450家族2亚家族C成员9)(图6C、D)。
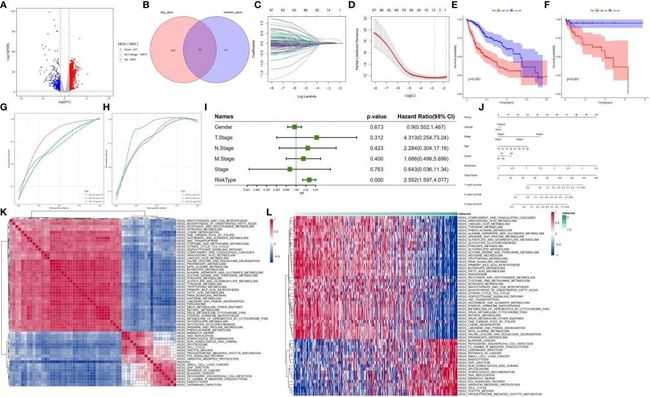
图6 在训练(TCGA-LIHC)和验证队列(GSE76427)中的HDG鉴定和验证
接下来,中位风险评分被用来将患者分为高风险组和低风险组。低风险组的患者显示出明显更高的总生存率,相比于高风险组的患者(p<0.001)(图6E)。在验证队列中应用这11个基因标记也表明,低风险组的患者比高风险组的患者有更长的总生存率(p<0.001)(图6F)。为了测试这11个基因标记的预后表现,作者生成了针对TCGA-LIHC样本的时间相关ROC曲线。结果显示,在测试队列中,1年、3年和5年生存的曲线下面积(AUC)值分别为0.8、0.7和0.7(图6G),在验证队列(GSE76427)中分别为0.8、0.8和0.87(图6H)。这些发现表明,这11个基因标记在两个队列中都具有良好的预后价值。对于临床病理特征与预后模型之间的关联分析,作者分析了TCGA-LIHC样本中的性别、TNM分期和风险评分。多元Cox回归分析结果显示,风险评分是肝细胞癌患者的一个显著独立预后因子(p<0.001)(图6I)。接下来,作者采用单样本GSEA方法,根据风险评分计算每个样本在175个通路上的得分,以确定相关的调控通路。然后,对每个通路与风险评分之间的相关性进行评估,相关性不低于0.3。结果显示,有39个通路与样本风险评分呈正相关,其中包括与癌症发展相关的通路;而与风险评分呈负相关的通路包括调节糖酵解/糖原合成、甘氨酸代谢、脂肪酸代谢、丝氨酸、苏氨酸、乙酰乙酸和二羧酸代谢等通路(图6K,L)。
7. MARCKSL1、SPP1、BSG、CCT3、LAGE3、KPNA2、SF3B4、GTPBP4、PON1、CFHR3和CYP2C9的相对RNA表达水平和蛋白质表达水平
根据不同上下调基因的初步趋势,为了进一步研究11个与预后相关的差异表达基因(MARCKSL1、SPP1、BSG、CCT3、LAGE3、KPNA2、SF3B4、GTPBP4、PON1、CFHR3和CYP2C9)在肝细胞癌(HCC)患者的高风险组和低风险组中的基因表达特征,作者进行了基因表达水平和风险评分之间的相关性分析。结果显示,除了PON1、CFHR3和CYP2C9之外,所有八个基因的表达水平与风险评分呈正相关。同时,为了对高风险和低风险基因进行分类,作者可以从11个预后基因的森林图中看到,MARCKSL1、SPP1、BSG、CCT3、LAGE3、KPNA2、SF3B4和GTPBP4的风险比均大于1,表明这8个基因可能是不良预后因子,属于高风险基因,而PON1、CFHR3和CYP2C9均小于1,表明这三个基因可能是预后较好的因子。通过qPCR比较了正常人肝细胞系MIHA和HCC细胞系HCC-LM3(高转移性HCC细胞)以及HepG2(低转移性HCC细胞)中MARCKSL1、SPP1、BSG、CCT3、LAGE3、KPNA2、SF3B4、GTPBP4、PON1、CFHR3和CYP2C9的RNA表达。发现与正常人肝细胞相比,人肝癌细胞中CYP2C9、PON1和CFHR3的表达较低,而MARCKSL1、SPP1、BSG、CCT3、LAGE3、KPNA2、SF3B4、GTPBP4的表达较高(未配对t检验,p<0.01)(图7A)。图7B显示了HepG2和HCC-LM3中CYP2C9和PON1的蛋白表达水平下调,与MIHA相比。同时,CPTAC数据库分析结果显示,与癌旁正常组织相比,肿瘤组织中PON1、CFHR3和CYP2C9的蛋白表达较低,而其他基因的表达较高(图7C)。
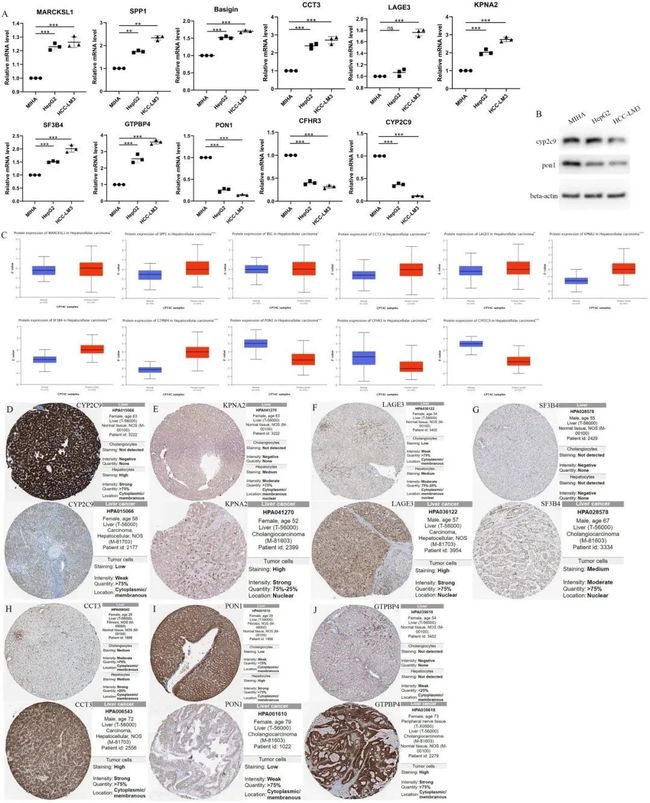
图7 预后相关差异表达基因的相对RNA表达水平和蛋白质表达水平
此外,来自HPA数据库的免疫组化分析证实了HCC组织中KPNA2、LAGE3、SF3B4、CCT3和GTPBP4蛋白表达较高,而CYP2C9和PON1蛋白表达较低(图7D-J)。
8.使用CMap对高风险群体进行风险模型的药物敏感性分析和潜在化合物的靶向作用
为了确定风险对临床实践的影响,作者使用“oncoPredict”软件包评估了高风险组和低风险组中多种化疗药物的IC50值。这项分析发现了123种在统计学上显著(p < 0.01)的药物。结果显示,阿法替尼、达沙替尼、5-氟尿嘧啶、拉帕替尼、SCH772984和塞地利尼在高风险组中的IC50值较低,相比低风险组,这意味着高风险组的患者可能更能从这些药物中获益。相反,JQ1、AT13148、阿西替尼、AZ960、AZD1208和伊立替康在低风险组中的IC50值较低,这意味着低风险患者可能更能从上述化疗药物中获益(图8A)。
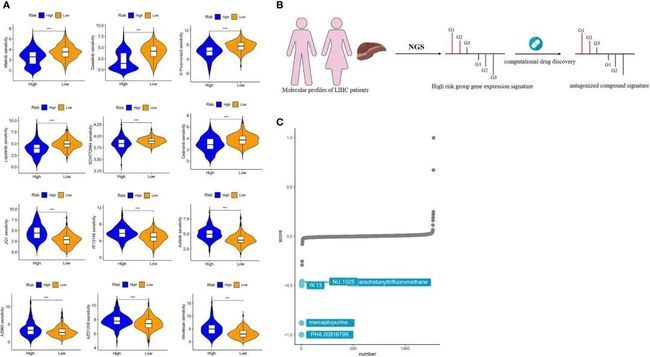
图8 药物敏感性分析和风险模型的靶化合物筛选
单细胞测序策略是构建特定于个体细胞类型的疾病特征的强大工具,而CMap为研究人员提供了前所未有的便利,将药物、基因和疾病的三元关系紧密联系在一起,而在这种情况下,对深入理解的需求不高,因为该方法不需要提前提供详细的作用机制或药物靶点来预测治疗潜力。因此,通过将高分辨率的单细胞测序策略与CMap相结合,作者能够根据个体细胞水平的表达特征直接针对有效的治疗药物,并为筛选潜在疾病药物提供更准确的预测。
作者采用了基于“特征逆转”的计算药物发现策略来识别在CMap数据库中具有逆转风险的药物,利用了大量的数据(图8B)。从高风险组和低风险组中提取了具有最高折叠变化的前300个基因进行XSum分析。CMap分析的结果显示,有几种化合物的基因表达模式与高风险组特异性相反,较低的CMap分数表示更高的干扰能力。PHA.00816795、巯基嘌呤、W.13、NU.1025和花生四氟甲烷是五种潜在有价值的小分子药物候选物,因为它们在候选物排名中位居前五(图8C)。在这些候选物的前三名中,巯基嘌呤是一种常见的化疗药物,通过干扰细胞分裂或DNA合成产生抗癌效果。余等人通过整合多种数据来定义基因类型,考虑基因变化对HCC的影响以及药物与HCC之间的正负关系,获得了与HCC相关的五种药物。在这些药物中,巯基嘌呤是一种潜在的抗肝细胞癌药物。
总结
总之,本研究在单细胞水平上确定了与代谢变化显著相关的预后基因,并探索了这个细胞亚群的异质性及其与肿瘤微环境中其他细胞的相互关系。建立并验证了一个用于预测肝细胞癌患者生存期的预后模型,结果显示出良好的预测能力。此外,评估了高风险组和低风险组之间的化疗敏感性差异,并预测了可能逆转风险评分的五种潜在药物。这些结果深入了解了肝细胞癌的代谢特征。此外,通过比较由肝恶性细胞和正常肝细胞构建的与肿瘤相关的基因,可以明确潜在预后生物标志物的特征。以上研究可能有助于个体化治疗的新策略的制定。