DAEMKL:基于多核学习的深度自动编码器预测miRNA与疾病的关联(IEEE Transactions on Neural Networks and Learning Systems)
DAEMKL:Predicting miRNA–Disease Associations Through Deep Autoencoder With Multiple Kernel Learning(发表于IEEE Transactions on Neural Networks and Learning Systems)
Predicting miRNA-Disease Associations Through Deep Autoencoder With Multiple Kernel Learning | IEEE Journals & Magazine | IEEE Xplore https://ieeexplore.ieee.org/document/9635589
https://ieeexplore.ieee.org/document/9635589![]()
![]()
目录
摘要(Abstract)
1.引言(Introduction)
2.材料和方法(MATERIALS AND METHODS)
A.已证实的人MDAs(已证实的人MDA )
B. MiRNA Space
C. Disease Space
D.多核学习(Multiple Kernels Learning)
E.特征表示的回归模型(Regression Model for Feature Representation)
F.深度自动编码器
G.DAEMKL
3.结果与讨论(RESULTS AND DISCUSSION)
A. 实现细节(Implementation Details)
B.性能评价(Performance Evaluation)
C.多核学习的效果(Effect of Multiple Kernels Learning)
D. 案例研究(Case Studies)
4. 结论(conclusion)
摘要(Abstract)
确定microRNA(miRNA)-疾病关联是预防、诊断和治疗复杂疾病的重要组成部分。 然而,湿实验识别MDAs效率低且昂贵。 因此,建立可靠有效的数据集成模型预测MDAs具有重要意义。 本文提出了一种基于多核学习的深度自动编码器(DAEMKL)的MDAs预测方法。 首先,DAEMKL在miRNA空间和疾病空间分别应用多核学习(MKL)构建miRNA相似度网络和疾病相似度网络。 然后,通过回归模型从miRNA相似度网络和疾病相似度网络中学习每个疾病或miRNA的特征表示。 然后将整合后的miRNA特征表示和疾病特征表示输入到深度自动编码器(DAE)中。 此外,通过重构误差预测新的MDAs。 最终,AUC的结果表明,DAEMKL实现了出色的性能。 此外,三种复杂疾病的案例研究进一步证明,DAEMKL具有卓越的预测性能,能够发现大量潜在的MDAs。 总的来说,我们的方法DAEMKL是一种有效的MDA识别方法。
索引术语-深度自动编码器(DAE),特征表示,microRNA(miRNA)-疾病关联(MDAs),多核学习(MKL)。
1.引言(Introduction)
mIcroRNAs(miRNAs)是一组长度约为22个核苷酸的非蛋白编码核糖核酸(RNAs),是多种生物过程的关键调节因子[1]-[3]。 miRNA及其靶mRNA的生物合成和功能障碍可能导致各种疾病[4]。 研究表明,miRNAs与乳腺癌、胰腺癌、淋巴瘤等多种恶性疾病的预防、诊断和治疗密切相关[5]、[6]。 因此,研究miRNA与疾病的关联在生物医学中具有重要意义。 然而,传统的生物实验方法鉴定MDAs费时费力,需要设备。 因此,越来越多的研究者转向生物信息学的方法来分析和预测新的MDAs。
以往预测MDAs的计算方法主要归纳为三类[7]。 第一类是基于相似性度量的方法,基于相似疾病更有可能与相似的miRNAs相关,反之亦然的假设。 Xuan等人[8]提出了预测MDAs的加权k最相似邻居。 该方法综合疾病术语和疾病表型相似性,可有效预测MDAs。 陈等人[9]提出了一种基于随机游走重启的全局网络相似性度量方法,用于预测新的MDAs(RWRMDA)。 不久之后,施等人[10]提出了另一种新的随机游走方法来预测MDAs。 该方法将疾病基因和miRNA靶基因映射到蛋白质相互作用网络中,利用随机游走法预测MDAs。 You等人[11]提出了一种基于路径的预测MDAs(PBMDAs)的方法。 PBMDA构造了一个异构网络,然后采用深度优先搜索方法预测新的MDAs。 由于考虑了网络拓扑结构,PBMDA取得了很好的预测效果。 Xiao等人[12]提出了一种利用自适应多源多视图潜在特征识别新MDAs(M2LFL)的新方法。 M2LFL采用自适应联合图正则化项将MiRNAs的与疾病的manifold structures结合起来。 最后,该方法取得了较好的预测效果。
第二类是基于机器学习的方法。 近年来,机器学习在各个领域都很受欢迎。 多种预测MDAs的机器学习模型也得到了众多学者的认可。 2013年,Jiang等人[13]提出了一种基于支持向量机(SVM)的方法。 他们从每个MDA数据中提取特征,用于训练SVM分类器,以挖掘未知MDAs的潜在信息。 Chen等人 [14]实现了集成学习和链接预测来发现MDAs(ELLPMDAs)。ELLPMDA通过集成学习综合了三种算法得到的预测结果。 此外,Chen等人[15]提出了另一种方法,即用归纳矩阵补全法预测潜在的MDAs(IMCMDAs)。IMCMDA的动机是使用已知的MDAs信息、miRNA相似性和疾病相似性信息来补充缺失的关联。还采用了一些矩阵分解方法来预测MDAs。 Xiao等人提出了一种基于图正则化非负矩阵分解(GRNMF)的MDAs预测框架。 最近,Gao等人[16]发展了另一种发现新MDAs的矩阵分解方法,称为基于最近谱的协同矩阵分解(NPCMF)。 NPCMF考虑了相似度网络的邻居信息,对miRNA相似度矩阵和疾病相似度矩阵施加了最近的谱,取得了良好的预测效果。
第三类是基于深度学习的方法。 随着深度学习方法在自动驾驶、人脸识别等领域的成功应用[17]-[19],研究人员开始开发一些关于MDA预测的深度学习方法。 例如,chen等人[20]提出了一种基于深度信念网络的MDAs预测方法(DBNMDAs)。 DBNMDA首先从所有miRNA-疾病对中提取特征来训练受限玻尔兹曼机器,然后选择与正样本数量相同的负样本来调整深度信念网络。 此外,Ji等人[21]提出了一种基于自动编码器的预测新MDAs(AEMDAs)的计算方法。 AEMDA通过深度学习算法训练了三种模型,包括miRNA模型、疾病模型、深度自动编码器(DAE)模型。 AEMDA首先训练miRNA模型和疾病模型获得特征表示,然后输入AutoEncoder完成MDAS的预测。 最后,AEMDA取得了较好的预测性能。
尽管上述方法取得了优异的性能,但它们仍然面临着一些局限性。 一方面,现有数据库中只有少量已知关联作为阳性样本,其余的样本都是未知关联,因此没有经过验证的阴性样本。 因此,一些有监督的机器学习算法很难训练出可靠的预测模型。 另一方面,在构建miRNA相似度核和疾病相似度核的过程中,大量方法只是简单地将高斯核与miRNA功能相似度核和疾病语义相似度核结合起来[16]。
为了克服上述不足,本研究通过多核学习的深度自动编码器(DAEMKL)开发了一个更完整的预测MDAs的深度学习框架。 在本研究中,DAE用于学习潜在的疾病相关miRNAs的特征,而不需要阴性样本。 最重要的是,DAEMKL集成了来自miRNA特征空间和疾病特征空间的多种信息。 几个核分别由miRNA和疾病空间构建。 然后将多核学习(MKL)应用于miRNA空间和疾病空间,构造miRNA相似度网络和疾病相似度网络。 基于多个不同信息源的MKL可以包含更多的先验信息,有利于捕捉深度交互。 对于每种疾病或 miRNA,其特征表示是通过回归模型从 miRNA 相似性网络和疾病相似性网络中学习得到的。MIRNA和疾病特征表示以类似的方式学习,以确保后续学习过程中数据的兼容性。 之后,将整合后的miRNA特征表示和疾病特征表示输入DAE。 最后,DAEMKL对输入数据进行重构,根据重构误差预测新的MDA。 因此,采用五折交叉验证(5-CV)和全局留一交叉验证(LOOCV)来验证预测性能。 此外,还进行了三种疾病的病例研究,以验证预测的新MDAS是否已被记录。 DAEMKL充分利用了先验知识和特征信息的兼容性,取得了良好的预测性能,挖掘出了更多未知的MDA。 本文的主要贡献概括如下。
1)开发了一个完整的基于深度学习的端到端预测框架,能够有效识别新的MDAs。
2)DAE作为预测器,可以有效地从已知的MDAs中学习特征信息,而不需要负样本。
3)将MKL引入到预测框架中,学习多源信息,使miRNAs和疾病的信息更加丰富。
本文的组织方式如下:第二节展示了数据集和用于预测MDAs的DAEMKL模型。 第三节介绍了实验结果和评价方法。 此外,我们的模型也与其他先进的方法进行了比较。 第四节对全文进行了总结。
2.材料和方法(MATERIALS AND METHODS)
A.已证实的人MDAs(已证实的人MDA )
已知的MDAs是通过人类小RNA疾病数据库(HMDD)V2.0[22]和HMDD V3.2[23]获得的。 HMDD是一个由研究人员手工收集的MDA数据库。 每个记录的MDA都包含详细的信息,不仅包括miRNA和疾病名称,还包括相关说明和参考文献的PubMed ID。 第一个数据集(D1)被认为是预测MDAs的金标准数据集,包括495个miRNA,383个疾病和5430个已证实的MDA。 第二个数据集(D2)也广泛应用于MDA预测[24],[25],包含550个miRNA,328个疾病和6088个MDA。 使用HMDD V3.2验证模型预测的新MDA是否被记录。 考虑到模型的适用性,DAEMKL模型和其他先进的方法使用D1和D2进行评估,以验证DAEMKL模型的性能。
B. MiRNA Space
为了表征不同信息源的miRNAs的特征,构造了miRNAs的三种相似核,包括miRNAs功能相似度、高斯互作谱(GIP)核相似度和Jaccard相似度。
miRNA功能相似性从MISIM数据库中获得[26]。 MISIM数据库(http://www.cuilab.cn/files/images/cuilab/misim.zip)由Wang等人开发。 基于功能相似的miRNAs更可能与表型相似的疾病相关。 由此构造了miRNA功能相似矩阵![]() ,其中
,其中![]() 表示miRNA i 和miRNA j 的功能相似度。
表示miRNA i 和miRNA j 的功能相似度。
为了获得miRNA空间的网络相似度,根据前人的研究[27]计算了miRNA的GIP核相似度![]() 。 miRNA i 和miRNA j 之间的GIP核相似度定义为
。 miRNA i 和miRNA j 之间的GIP核相似度定义为
![]()
其中![]() 是miRNA i 和miRNA j 的关联谱,即分别是关联矩阵的第i列和第j列。 此外,θm是内核带宽的可调参数,定义如下:
是miRNA i 和miRNA j 的关联谱,即分别是关联矩阵的第i列和第j列。 此外,θm是内核带宽的可调参数,定义如下:
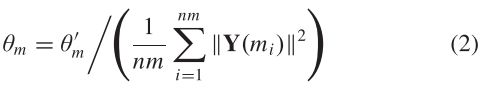
其中nm是miRNAs的数目,θm通常取为1[27]。
Jaccard相似度在文本挖掘和数据聚类等任务中得到了广泛的应用。 在这里,我们采用它来度量miRNAs之间的相似性。 公式如下:

C. Disease Space
在疾病空间中,相似核也包括三类:疾病语义相似度、GIP核相似度和Jaccard相似度。
语义相似度被认为是衡量疾病之间关系的可靠指标[28]。 根据前人的研究[26],基于医学主题词(Mesh)数据库开发了有向无环图(DAG)来计算疾病的语义相似度![]() 。DAG中有大量的点和有向边,其中每个点是一个疾病,每个有向边表示疾病之间的关系。
。DAG中有大量的点和有向边,其中每个点是一个疾病,每个有向边表示疾病之间的关系。
根据相同的计算规则,GIP内核相似度![]() 和Jaccard相似度
和Jaccard相似度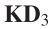 如下所示:
如下所示:
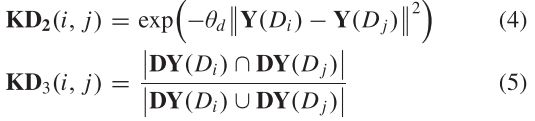
其中![]() 是疾病I和疾病J的关联谱,分别表示关联矩阵的第i行和第j行。
是疾病I和疾病J的关联谱,分别表示关联矩阵的第i行和第j行。 ![]() 表示关联图谱中与疾病 j 相关的miRNAs集合。
表示关联图谱中与疾病 j 相关的miRNAs集合。
D.多核学习(Multiple Kernels Learning)
考虑到单个相似核或miRNA功能相似矩阵与GIP核相似矩阵的简单组合不能表示miRNA之间的相似信息,采用多核学习(MKL)来获得最优核[29]。 以图为例说明了MKL的过程 1. MKL通过一个优化算法计算每个核的权重,从而得到一个miRNA的集成核。

miRNAs的最优核![]() 定义为
定义为

其中![]() 是miRNA核的最优权值,N是miRNA核的个数。 MKL的学习过程是利用理想核对集成核进行优化。 公式如下:
是miRNA核的最优权值,N是miRNA核的个数。 MKL的学习过程是利用理想核对集成核进行优化。 公式如下:

其中,![]() 表示Frobenius范数,
表示Frobenius范数,![]() ,β表示正则化参数。 上述优化问题可计算如下:
,β表示正则化参数。 上述优化问题可计算如下:

同样,通过上述MKL算法也可以计算出疾病核的权重![]() 。 疾病的最终最优核
。 疾病的最终最优核![]() 可得到如下:
可得到如下:

E.特征表示的回归模型(Regression Model for Feature Representation)
受 Ji 等人的工作启发。 [21]中,通过回归模型从最终的最优相似度核学习miRNA特征表示和疾病特征表示。 在miRNA空间中,对于每一个miRNA,我们构造了一个高维向量![]() 来表示高维空间中的特征。 所有高维向量
来表示高维空间中的特征。 所有高维向量![]() 的集合构成miRNA的特征矩阵
的集合构成miRNA的特征矩阵 。 特征矩阵
。 特征矩阵![]() 可以表示如下:
可以表示如下:
![]()
其中![]() 表示i th miRNA的向量,kM 表示
表示i th miRNA的向量,kM 表示![]() 的大小,nm表示miRNA的数目。 在回归模型的训练过程中,特征矩阵随机初始化。
的大小,nm表示miRNA的数目。 在回归模型的训练过程中,特征矩阵随机初始化。
此外,余弦相似度被用作测量![]() 之间距离的一种可靠有效的方法。 回归模型采用最优相似核
之间距离的一种可靠有效的方法。 回归模型采用最优相似核![]() 作为 ground-truth 标签。 但是,余弦相似度计算值的取值范围为[-1,1],在
作为 ground-truth 标签。 但是,余弦相似度计算值的取值范围为[-1,1],在![]() 中的相似度值是非负的。 因此,余弦相似度得分的范围在0和1之间调整如下:
中的相似度值是非负的。 因此,余弦相似度得分的范围在0和1之间调整如下:

其中![]() 分别是miRNA i 和miRNA j 的特征表示向量,而
分别是miRNA i 和miRNA j 的特征表示向量,而![]() 表示这两个miRNA的距离度量。 然后,构造miRNA回归模型来学习miRNAs的特征表示,该模型使
表示这两个miRNA的距离度量。 然后,构造miRNA回归模型来学习miRNAs的特征表示,该模型使![]() 和最优相似核
和最优相似核![]() 最小化。 公式如下所示:
最小化。 公式如下所示:

其中![]() 是训练样本的数目。 在训练过程中,采用平方损失作为反向传播的标准,并采用随机梯度下降(SGD)方法更新特征矩阵。 经过多次训练,得到了可靠的miRNA特征矩阵。
是训练样本的数目。 在训练过程中,采用平方损失作为反向传播的标准,并采用随机梯度下降(SGD)方法更新特征矩阵。 经过多次训练,得到了可靠的miRNA特征矩阵。
类似地,疾病![]() 的可靠特征表示可以通过回归训练。 公式定义如下:
的可靠特征表示可以通过回归训练。 公式定义如下:

其中 nd 为疾病数,KD 表示向量大小。 在训练过程中,我们将 kD=kM 设置为0.5。 CD是疾病的余弦相似度量,![]() 也是训练样本数。
也是训练样本数。
F.深度自动编码器
一个典型的自动编码器被定义为一个无监督学习模型,其目的是重建输入特征[30],[31]。 近年来,DAE是一种常用的深度学习方法,在特征提取、模式识别等领域得到了广泛的应用[32]-[34]。 因为数据集中只有一小部分关联是被证实的,我们开发了一种基于DAE的半监督学习方法。 我们的模型分为编码和解码两个部分。 在编码部分,将miRNA的高维特征向量![]() 和疾病的高维特征向量
和疾病的高维特征向量![]() 串联起来作为自动编码器的输入
串联起来作为自动编码器的输入![]() 。 然后编码后得到潜在变量
。 然后编码后得到潜在变量![]() 。 之后,DAE在解码部分根据潜在变量
。 之后,DAE在解码部分根据潜在变量![]() 输出重构
输出重构![]() 。
。
在训练过程中,已知的MDAs被认为是可观察的样本。 DAE的训练样本![]() 通过miRNA的特征向量
通过miRNA的特征向量![]() 和疾病的特征向量
和疾病的特征向量![]() 连接起来。 训练样本
连接起来。 训练样本![]() 定义如下:
定义如下:
![]()
其中![]() 是第i个训练样本。 对于样本
是第i个训练样本。 对于样本![]() ,编码过程的作用是将样本
,编码过程的作用是将样本![]() 的高维特征转换为潜在变量
的高维特征转换为潜在变量![]() 。 编码过程定义如下:
。 编码过程定义如下:

其中![]() 是编码器中隐藏层的数目,本文将
是编码器中隐藏层的数目,本文将![]() 设置为2。
设置为2。 ![]() 表示
表示![]() ,
,![]() 是第
是第![]() 隐层特征表示。
隐层特征表示。![]() 表示权重矩阵,
表示权重矩阵,![]() 表示偏差。 另外,非线性激活函数
表示偏差。 另外,非线性激活函数![]() 是获取表示一种可靠的方法。
是获取表示一种可靠的方法。
解码器的输出是根据潜在变量![]() 重构的特征表示
重构的特征表示![]() ,解码器定义如下:
,解码器定义如下:

其中![]() 是编码器输出的潜变量
是编码器输出的潜变量![]() ,
,![]() 表示权重矩阵,
表示权重矩阵,![]() 表示第
表示第![]() 层中的偏置。
层中的偏置。 ![]() 是译码器对输入
是译码器对输入![]() 输出的重构,tanh(·)是非线性激活函数。
输出的重构,tanh(·)是非线性激活函数。
最后分两部分计算了损失函数。 第一部分是所有重建误差的平方损失之和,第二部分是一个正则化项。 因此,DAEMKL的损失函数可以表示为

其中![]() 是训练样本数,λ是超参数,
是训练样本数,λ是超参数,![]() 是Jacobian的正则化。 在训练的每个阶段,DAEMKL通过最小化
是Jacobian的正则化。 在训练的每个阶段,DAEMKL通过最小化![]() 的损失来更新模型中的所有参数。
的损失来更新模型中的所有参数。
G.DAEMKL
DAEMKL 的整个流程图如图2所示。我们的模型框架可以用四个步骤来描述。首先,采用 MKL 分别获得 miRNA 空间和疾病空间的最优核。其次,通过回归模型从miRNA空间和疾病空间的最优核学习每个miRNA和每个疾病的特征表示。 第三,将整合后的miRNA特征表示和疾病特征表示输入DAE。 最后,通过重构误差预测新的MDA。

3.结果与讨论(RESULTS AND DISCUSSION)
A. 实现细节(Implementation Details)
DAEMKL通过以下方案进行训练。 首先,以均方损失作为误差反向传播准则,采用SGD方法对回归模型进行端到端的训练。 此外,采用Adam算法对回归模型进行优化。 经过60个epoch,得到了M和D的两个特征表示。 此外,DAE是用M、D和已知的MDAs训练的,DAE通常在50-100个epoch后达到收敛。 我们采用M和D的串联向量作为输入,因此输入层有![]() 神经元。 编码器的隐层节点分别设置为
神经元。 编码器的隐层节点分别设置为![]() ,解码器的隐层节点分别设置为
,解码器的隐层节点分别设置为![]() 和
和![]() 。
。
此外,DAEMKL的初始学习速率![]() 、DAEMKL的训练epoch
、DAEMKL的训练epoch![]() 、向量大小
、向量大小![]() 和
和![]() 等超参数也影响DAEMKL的性能。 我们在数据库D1上使用这些参数的不同组合对DAEMKL进行训练,这些参数的范围为
等超参数也影响DAEMKL的性能。 我们在数据库D1上使用这些参数的不同组合对DAEMKL进行训练,这些参数的范围为![]() ∈[512,1024,2048,4096]、
∈[512,1024,2048,4096]、![]() ∈[50,100,150,200]和
∈[50,100,150,200]和![]() ∈[1,1e-1,1e-2,1e-3,1e-4]。 通过经验调整参数,我们在下面的实验中为DAEMKL设置
∈[1,1e-1,1e-2,1e-3,1e-4]。 通过经验调整参数,我们在下面的实验中为DAEMKL设置![]()
![]() 。
。
B.性能评价(Performance Evaluation)
在实验中,采用5-CV和全局LOOCV对DAEMKL的预测性能进行了评估。 对于5-CV,已知的MDAs被随机分为五个子部分。 每个子部分依次作为测试样本,其余四个子集作为训练集。 AUC的值是接收机工作特性(ROC)曲线下的面积。 AUC的值在0到1之间,其中0.5表示随机性能。 AUC值小于0.5表示预测结果无意义。 为了防止结果的偶然性,在每一轮中,都使用训练样本对DAEMDA进行训练。 然后我们计算剩余样本和未确认样本的重建误差。 跑50次结果的平均为最后的结果。
在应用5-CV和Global LOOCV对我们的方法进行评估时,我们还将DAEMKL与五种先进的MDA预测方法:IMCMDA[15]、PBMDA[11]、EGBMMDA[35]、NPCMF[16]和AEMDA[21]进行了比较。 为了将DAEMKL与最先进的方法进行比较,我们在两个数据集D1和D2上实现了5-CV,并在数据集D1上实现了全局LOOCV。 表一和图 3给出了DAEMKL的详细结果和比较方法。结果表明,DAEMKL 在 D1上获得了5-CV 为0.9583的 AUC 和全局 LOOCV 为0.9671的 AUC,两者与其他模型相比均排名第一。因此,DAEMKL 的性能明显优于其他五种最先进的方法。


C.多核学习的效果(Effect of Multiple Kernels Learning)
在本研究中,多核学习被引入到我们的预测框架中,这有助于分别在miRNA空间和疾病空间提取高维特征。 通过比较DAEMKL和不使用MKL的模型(使用单个内核或平均内核)的性能,研究了MKL的性能。 平均核将GIP核与miRNA功能相似度核和疾病语义相似度核相结合。图4和5显示了不同核在5-CV中的性能比较。 实验标记K1为![]() ,或K2为
,或K2为![]() ,或K3为
,或K3为![]() 。
。


D. 案例研究(Case Studies)
为了进一步证明我们的方法的预测性能,并证实 DAEMKL 在现实情况下获得的预测结果的可靠性和合理性,我们对淋巴瘤,结肠新生物和胰腺肿瘤进行了病例研究。在实验中,D1数据集被用来训练我们的模型来考虑疾病的优先候选 miRNA。然后,选择与疾病相关的前30个潜在的miRNAs,并用MDA数据库的最新版本DBDEMC V2.0[36]和HMDD V3.2[37]对预测的miRNAs进行验证。
结肠肿瘤是胃肠道最常见的恶性肿瘤之一。 2018年中国肿瘤统计数据显示,我国结肠癌发病率和死亡率分别居所有恶性肿瘤的第三位和第五位。 为了评估DAEMKL的预测效果,我们选择结肠肿瘤作为我们的第一个病例研究。 如表II所列,96.67%的前30个miRNA与结肠肿瘤的关联已被这两个数据库验证。 在这30个新的关联中,60%的关联被DBDEMC V2.0和HMDD V3.2数据库确认。 许多研究证明,结肠肿瘤的发生发展与许多miRNAs密切相关。 例如,zHANG等人 [38]发现miR-21、miR-17和miR-19a促进结肠癌细胞的增殖和侵袭。

胰腺癌是一种高度致命的恶性肿瘤,存活率非常低。 大多数患者被诊断为晚期疾病,因此可用的治疗方法不太有效[40]。 因此,在胰腺肿瘤中识别潜在的miRNAs可以改善患者的预后。 本研究选取了前30个预测的胰腺癌相关miRNAs进行分析。 在筛选出的30个miRNA与胰腺癌的关联中,96.67%的关联可以在DBDEMC V2.0或HMDD V3.2数据库中找到证据。 另外,53.3%的胰腺癌与miRNA相关,只有miR-219未被证实。然而,Lahdaoui等人[39]发现 miR-219在胰腺癌细胞系中的过度表达导致细胞增殖和迁移减少。 具体详情载于表三。

淋巴瘤是一种起源于淋巴造血系统的恶性肿瘤,可累及身体的任何器官[40]。 2012年,全球约有386000名非霍奇金淋巴瘤患者和66000名霍奇金淋巴瘤患者,占癌症总数的3.2%[41]。 研究表明,miRNA生物标志物的存在对淋巴瘤的早期发现具有重要作用。 例如,张等人。 [42]在套细胞淋巴瘤中检测到miR-15a和miR-16下调。 此外,Li等人。 [43]发现miR-21水平的表达可作为弥漫性大B细胞淋巴瘤预后的一个新的生物标志物。 在本文中,我们将验证两个数据库是否记录了预测的MDA。 DAEMKL实现后,发现前30个新的miRNA与淋巴瘤的关联中有28个得到了DBDEMC V2.0和HMDD V3.2数据库的验证。 尽管两个数据库都证实了只有两种新的 miRNA,但我们在 HMDD v3.2中发现了相对较少的淋巴瘤记录。因此,挖掘更多与淋巴瘤相关的潜在 miRNA 对于淋巴瘤的预防和治疗具有重要意义。具体结果列于表四。

4. 结论(conclusion)
研究表明miRNAs与许多复杂疾病有关。 识别与疾病相关的潜在miRNAs对于了解疾病发病机制和疾病治疗具有重要意义。 本文提出了一种基于多核学习的DAE预测框架,用于预测新的MDA。 第一步是将MKL应用于miRNA空间和疾病空间,构造miRNA相似网络和疾病相似网络。 然后,对每个疾病或miRNA通过回归模型从miRNA相似度网络和疾病相似度网络中学习其特征表示。 最值得注意的是,集成的miRNA特征表示和疾病特征表示被输入到DAE中。此外,通过重构误差对新型 MDA 进行了预测。最后,将DAEMKL算法与5-CV中的五种最先进的方法进行了比较,结果表明,DAEMKL算法在预测MDAS方面具有良好的性能。 此外,还对三种复杂疾病进行了仿真实验,进一步验证了DAEMKL的性能。 因此,多种性能评价指标的结果表明,DAEMKL比现有的其他方法表现出了更好的预测性能。 它是识别潜在疾病相关miRNAs的可靠有效的深层模型。
总的来说,DAEMKL中也存在局限性。 首先,利用回归模型分别学习miRNA和疾病的特征表示。 因此,当数据库变得非常大时,模型的复杂性就不是很友好了。 此外,目前验证的MDAs的不足也可能影响我们的模型的预测能力。 在未来的工作中,我们将致力于开发更高效的特征表示方法。 此外,我们将尝试整合更多不同的数据库,以进一步提高我们的预测模型的性能。